




Саркоми м’яких тканин: етіологія, епідеміологія, класифікація
Коровін С.І., Остафійчук В.В., Палівець А.Ю., Кукушкіна М.М., Волков І.Б., Костюк В.Ю.
Резюме. Саркоми м’яких тканин — це група відносно рідкісних злоякісних новоутворень різної локалізації та гістологічної структури, об’єднаних спільним походженням із мезодермальних тканин. Розвиваючись з ембріональних мезенхімальних клітин, вони згодом диференціюються в клітини поперечносмугастих, гладких м’язів, жирову або фіброзну тканину. За даними різних авторів, саркоми м’яких тканин становлять 15% усіх злоякісних пухлин у дітей і лише 1-2,5% — у дорослих. Метою огляду є ретельний аналіз існуючої літератури з епідеміології, етіології та класифікації сарком м’яких тканин.
 Саркоми м’яких тканин (СМТ) становлять невелику частку серед злоякісних пухлин у дорослих — 1–2,5% у структурі усіх злоякісних новоутворень [2] та близько 15% — у дітей. Наразі налічують близько 70 гістологічних підтипів. СМТ за особливостями росту, клінічним перебігом і прогнозом належать до найбільш злоякісних пухлин людини. Вони становлять велику групу новоутворень різноманітного гістогенезу, що вирізняються строкатою клінічною картиною, частим рецидивуванням, мультицентричним ростом, ранньою появою віддалених гематогенних метастазів і несприятливим прогнозом [60].
Саркоми м’яких тканин (СМТ) становлять невелику частку серед злоякісних пухлин у дорослих — 1–2,5% у структурі усіх злоякісних новоутворень [2] та близько 15% — у дітей. Наразі налічують близько 70 гістологічних підтипів. СМТ за особливостями росту, клінічним перебігом і прогнозом належать до найбільш злоякісних пухлин людини. Вони становлять велику групу новоутворень різноманітного гістогенезу, що вирізняються строкатою клінічною картиною, частим рецидивуванням, мультицентричним ростом, ранньою появою віддалених гематогенних метастазів і несприятливим прогнозом [60].
Етіологія
Хоча у більшості підтипів сарком немає визначеної причини появи, є дані про вплив зовнішніх факторів у розвитку СМТ. Так, існує зв’язок із вірусними інфекціями. Наприклад, доведено причетність вірусу Епштейна — Барр до розвитку лейоміосаркоми [37] чи саркоми Капоші, асоційованої з імунодефіцитом людини і в основному пов’язаної із підвищенням захворюваності на інфекцію, спричинену вірусом імунодефіциту людини (ВІЛ) [35, 70].
Доведено вплив радіаційного випромінювання на розвиток дерматофібросаркоми [6, 39], трапляються випадки виникнення злоякісної фіброзної гістіоцитоми через 6–30 років у хворих, які одержали курс променевої терапії [4, 26].
Також доведено спадкову схильність до розвитку СМТ. Так, при спадковому аденоматозному поліпозі ризик виникнення агресивного фіброматозу становить від 7,5 до 16% [13, 22, 47, 48]. При спадковій ретинобластомі частота розвитку лейоміосаркоми досягає 78% [32, 33]. Нейросаркома розвивається за наявності спадкового нейрофіброматозу з частотою 5%. При синдромі Лі — Фраумені ризик виникнення фібросаркоми та поліморфно-клітинної саркоми варіює від 12 до 21% [31, 42, 51]. Доведено розвиток астроцитоми та гліобластоми на фоні туберозного склерозу (хвороба Бурневіля) [59]. На тлі невоїдного базально-клітинного синдрому (синдром Горліна) зафіксовані випадки захворювання на фібросаркому щелепи [30]. Люди, які хворіють на синдром Вернера, мають ризик розвитку саркоми в 10% випадків [19–21].
Зв’язок між травматизацією та виникненням СМТ є досить дискутабельним; він є переважно сукупністю факторів одночасного травмування та виявлення пухлини пацієнтом [11].
Доведено підвищений ризик розвитку ангіосаркоми у людей, що працювали з арсеном [11].
Епідеміологія та демографічна характеристика
Як вже наголошувалося, загальносвітові показники захворюваності на СМТ становлять близько 15% усіх злоякісних пухлин у пацієнтів дитячого віку та лише 1–2,5% — у дорослих [2, 28].
Загальна частота виявлення СМТ підвищується з кожним роком у зв’язку з покращенням діагностики та підвищенням обізнаності лікарів первинної ланки. Також збільшення кількості хворих на СМТ пов’язано із зростанням частоти виявлення саркоми Капоші [35].
СМТ у дорослих в Європі загалом виникає рідко із прогнозованою частотою 4 випадки на 100 000 населення на рік [8]. Щорічна захворюваність на СМТ в США становить 10 390 випадків щороку із загальною смертністю 3680 осіб на рік [61]. Для порівняння, у 2013 р. в Україні зареєстровано 1760 випадків захворювання на злоякісні новоутворення м’яких тканин, рівень захворюваності при цьому становив 2,36 випадку на 100 000 стандартизованого населення. За даними Національного канцер-реєстру, у структурі захворюваності на злоякісні новоутворення в Україні захворюваність на СМТ займає 20-те місце серед чоловіків і 15-те — серед жінок. За частотою розвитку злоякісних пухлин у міських жителів СМТ займає 21-ше місце, у сільських — 24-те [1].
За 2000–2013 рр. в Україні зареєстровано 15 261 випадок захворювання. 45,0% усіх пацієнтів із СМТ становлять чоловіки і 55,0% — жінки. Найбільша частка припадає на хворих вікових груп 40–85 років і старше [1]. Найвищі показники захворюваності зареєстровано у старших вікових групах — 55–79 років — як у чоловіків, так і у жінок (рис. 1).
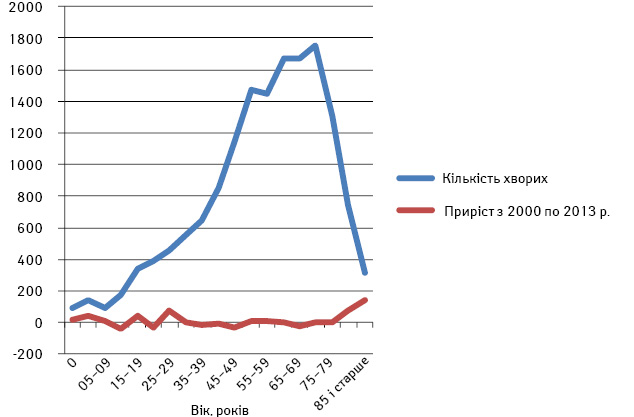
Рис. 1. Захворюваність у вікових групах
За вказаний період захворюваність на СМТ зросла на 2,5% серед жінок і знизилася на 1,8% — серед чоловіків.
Для проведення аналізу виживаності хворих зі злоякісними новоутвореннями м’яких тканин проаналізовано дані 8937 пацієнтів зі встановленим у 2000–2008 рр. діагнозом. 1-Річна кумулятивна виживаність цих хворих становила 54,9%. 5-Річна кумулятивна виживаність сягала 32,2%, серед міських жителів 5-річний рубіж пережили 33,9%, а серед сільських — тільки чверть (25,6%) пацієнтів. Така статистика пов’язана із неадекватним підходом до лікування пацієнтів, що зумовлено недостатньою обізнаністю онкологів стосовно зазначеної локалізації. За даними Національного канцер-реєстру України, за 2000–2013 рр. при виявленні захворювання відмічають високу частоту відсутності стадіювання хвороби в 53,1% випадків [1]. Така цифра зумовлена початковим лікуванням цієї когорти хворих у загальних хірургічних стаціонарах без участі онколога (рис. 2).
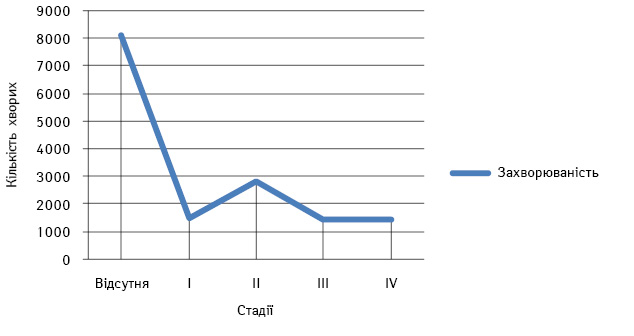
Рис. 2. Стадіювання хворих на СМТ за 2000–2013 рр.
Ураження СМТ виявляють практично на всіх ділянках тіла, але частіше пухлина виникає на нижніх кінцівках. Найчастішу локалізацію СМТ наведено в таблиці [57].
Таблиця. Локалізація СМТ
| Частини тіла | Частота ураження, % |
| Нижні кінцівки та таз | 43 |
| Заочеревинний простір та внутрішньочеревна порожнина | 20 |
| Верхні кінцівки та плечовий пояс | 18 |
| Тулуб | 10 |
| Голова і шия | 9 |
Класифікація
Згідно з класифікацією Всесвітньої організації охорони здоров’я 2013 р., до пухлин м’яких тканин належать доброякісні, злоякісні пухлини, а також пухлини із проміжним потенціалом злоякісності. Основними рубриками в зазначеній класифікації є пухлини жирової тканини (ліпосаркома), фібробластичні пухлини (фібросаркома), фіброгістіоцитарні пухлини, пухлини з гладких м’язів (лейоміосаркома), пухлини скелетних м’язів (рабдоміосаркома), периваскулярні пухлини, кістково-хрящові пухлини, стромальні пухлини, пухлини нервового стовбура, а також пухлини з нез’ясованим диференціюванням (синовіальна, епітеліоїдна, альвеолярна саркоми, саркома Юїнга). Наразі виділяють понад 70 гістологічних підтипів СМТ, що зумовлює варіабельність різних підходів до лікування. Трансформація і дедиференціювання доброякісної пухлини м’яких тканин в злоякісну трапляються рідко, виняток становлять нейрофіброми, які приблизно в 10% випадків можуть дедиференціюватися в злоякісну пухлину з оболонок периферичних нервів. Відмінності в частоті виникнення різних гістологічних підтипів СМТ зумовлені радше суб’єктивністю патоморфологів, а не географічною варіабельністю різних підтипів [14].
СМТ розвиваються із мезенхімальних тканин. Назву пухлина одержує залежно від типу тканини, з якої вона росте. Найбільш поширеними підтипами СМТ є злоякісна фіброзна гістіоцитома, гастроінтестинальна стромальна пухлина, ліпосаркома, лейоміосаркома, синовіальна саркома та злоякісна пухлина з оболонок периферичних нервів [9].
Злоякісна фіброзна гістіоцитома початково розвивається зі сполучної тканини, зокрема з клітин фібробластів [31, 42, 51]. Розрізняють декілька підтипів злоякісної фіброзної гістіоцитоми, такі як плеоморфна саркома, міксоїдна саркома, гігантоклітинна саркома, ангіоматоїдна злоякісна фіброзна гістіоцитома та її запальні варіанти [3, 15–17, 29, 43, 46, 50, 52]. Злоякісна фіброзна гістіоцитома є найбільш поширеною серед СМТ у дорослих та становить до 40% усіх СМТ [58]. Може розвиватися на будь-якій ділянці тіла та найбільш часто уражує нижні кінцівки; зазвичай хворіють люди літнього віку (≥50 років) із невеликим переважанням серед чоловіків. Характеризується агресивним ростом та ураженням прилеглих тканин. Є дослідження, що свідчать про розвиток злоякісної фіброзної гістіоцитоми на фоні ортопедичних імплантатів [64].
Гастроінтестинальна стромальна пухлина — найбільш розповсюджена пухлина шлунково-кишкового тракту мезенхімальної природи, становить 1–3% усіх гастроінтестинальних пухлин та займає 2-ге місце у структурі СМТ. До 2000-х років більше 70% гастроінтестинальних стромальних пухлин вважали доброякісними [7, 40, 49]. Тож більшість клінічних досліджень, проведених з метою вивчення цього підтипу СМТ до 2000 р., є малоінформативними. На думку науковців, гастроінтестинальні стромальні пухлини розвиваються з інтерстиціальних клітин Кахаля [41]. Найчастіше ураженню підлягає шлунок. Пік захворюваності припадає на вік 50–70 років з однаковою поширеністю у чоловіків та жінок [41]. 85–90% гастроінтестинальних стромальних пухлин дорослих є носіями онкогенних мутацій С-kit або PDGFRA [35, 36]. Відзначають розвиток цього підтипу СМТ при нейрофіброматозі 1-го типу та синдромі тріада Карнея [5, 45].
Ліпосаркома розвивається із жирових клітин, її частка становить близько 16–18% у загальній структурі СМТ [27, 55, 56, 62, 63]. Розрізняють декілька підтипів ліпосаркоми, найпоширенішими є: високодиференційована ліпосаркома, міксоматозна ліпосаркома, круглоклітинна ліпосаркома, поліморфно-клітинна ліпосаркома та недиференційована ліпосаркома [27, 44, 55, 56, 62, 63]. Пік захворюваності — 30–60 років, ураження фіксують переважно у чоловіків [10]. Цей вид СМТ може бути множинним та розвиватися на будь-якій ділянці тіла, але найпоширенішою локалізацією є заочеревинна клітковина [10, 56].
Лейоміосаркома — найбільш агресивна форма СМТ, що виникає із клітин гладких м’язів. З усіх СМТ близько 5–10% припадає на лейоміосаркому [23]. Лейоміосаркома м’яких тканин, як вважається, виникає з гладких м’язових клітин, що вистилають дрібні кровоносні судини. Лейоміосаркома може також розвиватися безпосередньо з гладких м’язів органів черевної порожнини, в тому числі шлунково-кишкового тракту і матки.
Лейоміосаркому м’яких тканин класично поділяють на три групи, що важливо для прогностичних та лікувальних цілей: лейоміосаркома «соматичних» м’яких тканин, лейоміосаркома шкіри, лейоміосаркома судинного походження [67]. Пік захворюваності — 50—60 років зі співвідношенням між жінками та чоловіками 2:1. Такий розподіл за статевим показником зумовлений впливом естрогенів на гладку м’язову тканину [18]. Доведеними причинами виникнення лейоміосаркоми є медикаментозна імуносупресія при трансплантації органів [66], набутий імунодефіцит людини при сукупності ураження ВІЛ та вірусом Епштейна — Барр [38]. Прогноз при лейоміосаркомі несприятливий — найнижчі показники виживаності серед усіх хворих на СМТ [35].
Синовіальна саркома розвивається зі сполучної тканини, зокрема фасції м’язів синовіальної оболонки суглобів, та становить 5–10% усіх СМТ [67]. Морфологічно виділяють 4 підтипи: волокнисту (схожа за будовою на фібросаркому), целюлярну (залозистоподібна), монофазну, біфазну.
Захворюваність на синовіальну саркому припадає на третю декаду життя та є однаковою у жінок і чоловіків — 1:1 [34, 67]. Причинами виникнення синовіальної саркоми у 90% є транслокація хромосом (X;18) (p11.2;q11.2) [53, 65]. Локалізуватися може на будь-якій ділянці тіла, але найчастіше уражаються нижні кінцівки, зокрема стопа [69].
Злоякісна пухлина з оболонок периферичних нервів — сама назва цього підтипу СМТ свідчить про розвиток захворювання із клітин оболонок периферичних нервів. Злоякісні пухлини з оболонок периферичних нервів становлять від 5 до 8% усіх СМТ [68]. Розвиток цього підтипу новоутворень у 10% випадків пов’язаний із нейрофіброматозом 1-го типу [24, 25, 62]. Описаний підтип пухлин включає у себе всі злоякісні пухлини, що розвилися з оболонок периферичних нервів: злоякісна неврома, нейрогенна саркома, нейрофібросаркома та злоякісна шваннома [12, 54]. Локалізуватися може на будь-якій ділянці тіла.
Висновки
СМТ — відносно рідкісна форма злоякісних новоутворень, які становлять 2,36 випадку на 100 000 населення і водночас є надзвичайно різноманітними за гістологічним типуванням, мають велику кількість етіологічних чинників, таких як генетична схильність, імунодефіцит людини, радіаційний фактор та ін. Характеризуються різнобарвною клінічною картиною, що є основною причиною діагностичних помилок та початку лікування таких хворих на пізніх стадіях пухлинного процесу, що зумовлює незадовільні результати терапії при цій нозологічній одиниці.
Список використаної літератури
1. Федоренко З.П., Михайлович Ю.Й., Гулак Л.О. та ін. (2015) Рак в Україні, 2013–2014. Захворюваність, смертність, показники діяльності онкологічної служби. Бюл. Національного канцер-реєстру України, 15: 104 с.
2. Шугабейкер П.Х., Малауэр М.М. (1996) Хирургия сарком м’яких тканей. Медицина, Москва, 440 с.
3. Al-Agha O.M., Igbokwe A.A. (2008) Malignant fibrous histiocytoma: between the past and the present. Arch. Pathol. Lab. Med., 132(6): 1030–1035.
4. Arlen M., Higinbotham N.L., Huvos A.G. et al. (1971) Radiation-induced sarcoma of bone. Cancer, 28: 1087–1099.
5. Boccon-Gibod L., Boman F., Boudjemaa S. et al. (2004) Separate occurrence of extra-adrenal paraganglioma and gastrointestinal stromal tumor in monozygotic twins: probable familial Carney syndrome. Pediatr. Dev. Pathol., 7(4): 380–384.
6. Brady M.S., Gaynor J.J., Brennan M.F. (1992) Radiation-associated sarcoma of bone and soft tissue. Arch. Surg., 127: 1379–1385.
7. Burkill G.J., Badran M., Al-Muderis O. et al. (2003) Malignant gastrointestinal stromal tumor: distribution, imaging features, and pattern of metastatic spread. Radiology, 226(2): 527–532.
8. Cancer MPact®. Kantar Health. Available from http://www.cancermpact.com. Accessed 04 10 2013.
9. Coindre J., Terrier P., Guillou L. et al. (2001) Predictive value of grade for metastasis development in the main histologic types of adult soft tissue sarcomas: a study of 1240 patients from the French Federation of Cancer Centers Sarcoma Group. Cancer, 91: 1914–1926.
10. Dei Tos A.P. (2000) Liposarcoma: new entities and evolving concepts. Ann. Diagn. Pathol., 4(4): 252–266.
11. Dirix L.Y., Vermeulen P., De Wever I., van Oosterom A.T. (1997) Soft tissue sarcoma in adults. Curr. Opin. Oncol., 9(4): 348–359.
12. Ducatman B.S., Scheithauer B.W., Piepgras D.G. (1986) Malignant peripheral nerve sheath tumors. A clinicopathologic study of 120 cases. Cancer, 57: 2006–2021.
13. Fallen T., Wilson M., Morlan B., Lindor N.M. (2006) Desmoid tumors — a characterization of patients seen at Mayo Clinic 1976–1999. Fam. Cancer, 5: 191–194.
14. Fanburg-Smith J.C., Dal Cin P. (2002) Angiomatoid fibrous histiocytoma. World Health Organization Classification of Tumors: Pathology and Genetics of Tumors of Soft Tissue and Bone. Lyon, France: IARC Press: 194–195.
15. Fletcher C.D. (1992) Pleomorphic malignant fibrous histiocytoma: fact or fiction? A critical reappraisal based on 159 tumors diagnosed as pleomorphic sarcoma. Am. J. Surg. Pathol., 16(3): 213–228.
16. Fletcher C.D., Gustafson P., Rydholm A. et al. (2001) Clinicopathologic re-evaluation of 100 malignant fibrous histiocytomas: prognostic relevance of subclassification. J. Clin. Oncol., 19(12): 3045–3050.
17. Fletcher C.D.M., Unni K.K., Mertens F. (2002) World Health Organization Classification of Tumours. Pathology and Genetics of Tumours of Soft Tissue and Bone. So-called fibrohistiocytic tumours. IARC Press, Lyon: 120–125.
18. Golden T., Stout A.P. (1941) Smooth muscle tumors of the gastrointestinal tract and retroperitoneal tissues. Surg. Gynecol. Obstet., 73: 784.
19. Goto M. (1997) Hierarchical deterioration of body systems in Werner’s syndrome: implications for normal ageing. Mech. Ageing Dev., 98: 239–254.
20. Goto M. (2000) Werner’s syndrome: from clinics to genetics. Clin. Exp. Rheumatol., 18(6): 760–766.
21. Goto M., Imamura O., Kuromitsu J. et al. (1997) Analysis of helicase gene mutations in Japanese Werner’s syndrome patients. Hum. Genet., 99(2): 191–193.
22. Gurbuz A.K., Giardiello F.M., Petersen G.M. et al. (1994) Desmoid tumours in familial adenomatous polyposis. Gut, 35: 377–381.
23. Gustafson P., Willen H., Baldetrop B. et al. (1992) Soft tissue leiomyosarcoma: a population-based epidemiologic and prognostic study of 48 patients, including cellular DNA content. Cancer, 70: 114.
24. Harbitz F. (1909) Multiple neurofibromatosis (von Recklinghausen’s disease). Arch. Int. Med., 3: 32–65.
25. Hosoi K. (1931) Multiple neurofibromatosis (von Recklinghausen’s disease). Arch. Surg., 22: 258–281.
26. Inoue Y.Z., Frassica F.J., Sim F.H. et al. (2000) Clinicopathologic features and treatment of postirradiation sarcoma of bone and soft tissue. J. Surg. Oncol., 75: 42–50.
27. Jelinek J.S., Kransdorf M.J., Shmookler B.M. et al. (1993) Liposarcoma of the extremities: MR an CT findings in the histologic subtypes. Radiology, 186: 455–459.
28. Jemal A., Tiwari R.C., Murray T. et al. (2004) Cancer statistics. CA Cancer J. Clin., 54: 8–29.
29. Kauffman S.L., Stout A.P. (1961) Histiocytic tumors (fibrous xanthoma and histiocytoma) in children. Cancer, 14: 469–482.
30. Khan A.A., Perveen S., Raza N., Ali Bukhari S.G. (2014) Gorlin — Goltz syndrome. J. Coll. Physicians Surg. Pak., 24(3): 171–173.
31. Kleihues P., Schauble B., zur Hausen A. et al. (1997) Tumors associated with p53 germline mutations: a synopsis of 91 families. Am. J. Pathol., 150: 1–13.
32. Kleinerman R.A., Schonfeld S.J., Tucker M.A. (2012) Sarcomas in hereditary retinoblastoma. Clin. Sarcoma Res., 2: 15.
33. Kleinerman R.A., Tucker M.A., Abramson D.H. et al. (2007) Risk of soft tissue sarcomas by individual subtype in survivors of hereditary retinoblastoma. J. Natl. Cancer Inst., 99: 24–31.
34. Ladanyi M., Antonescu C.R., Leung D.H. et al. (2002) Impact of SYT-SSX fusion type on the clinical behavior of synovial sarcoma: a multi-institutional retrospective study of 243 patients. Cancer Res., 62: 135–140.
35. Levi F., La Vecchia C., Randimbison L. (1999) Descriptive epidemiology of soft tissue sarcomas in Vaud, Switzerland. Eur. J. Cancer, 35: 1711–1716.
36. Mankin H.J., Casas-Ganem J., Kim J.I. et al. (2004) Leiomyosarcoma of somatic soft tissues. Clin. Orthop. Relat. Res., 421: 225–231.
37. McClain K.L., Leach C.T., Jenson H.B. et al. (1995) Association of Epstein — Barr virus with leiomyosarcomas in young people with AIDS. N. Engl. J. Med., 332: 12–18.
38. McClain K.L., Leach C.T., Lenson H.B. et al. (1995) Association of Epstein — Barr virus with leiomyosarcoma in young people with AIDS. New Eng. J. Med., 332: 19.
39. McLoughlin P.M., Girach M., Wood G.A. (1992) Dermatofibrosarcoma protuberans of the scalp. Br. J. Oral. Maxillofac. Surg., 30: 401–403.
40. Miettinen M., Lasota J. (2001) Gastrointestinal stromal tumors — definition, clinical, histological, immunohistochemical, and molecular genetic features and differential diagnosis. Virchows Arch., 438(1): 1–12.
41. Miettinen M., Lasota J. (2006) Gastrointestinal stromal tumors: review on morphology, molecular pathology, prognosis, and differential diagnosis. Arch. Pathol. Lab. Med., 130(10): 1466–1478.
42. Mitchell G., Ballinger M.L., Wong S. et al. (2013) High frequency of germline TP53 mutations in a prospective adult-onset sarcoma cohort. PLoS One, 8: e69026.
43. Murphey M.D. (2007) World Health Organization classification of bone and soft tissue tumors: modifications and implications for radiologists. Semin. Musculoskelet. Radiol., 11(3): 201–214.
44. Murphey M.D., Arcara L.K., Fanburg-Smith J. (2005) Imaging of musculosketal liposarcoma with radiologic-pathologic correlation. Radiol. Graphics., 25: 1371–1395.
45. Murphy J.D., Ma G.L., Baumgartner J.M. et al. (2015) Increased risk of additional cancers among patients with gastrointestinal stromal tumors: A population-based study. Cancer, 121(17): 2960–2967.
46. Nakayama R., Nemoto T., Takahashi H. et al. (2007) Gene expression analysis of soft tissue sarcomas: characterization and reclassification of malignant fibrous histiocytoma. Mod. Pathol., 20(7): 749–759.
47. Nieuwenhuis M.H., Casparie M., Mathus-Vliegen L.M. et al. (2011) A nationwide study comparing sporadic and familial adenomatous polyposisrelated desmoid-type fibromatoses. Int. J. Cancer, 129: 256–261.
48. Nieuwenhuis M.H., Lefevre J.H., Bulow S. et al. (2011) Family history, surgery, and APC mutation are risk factors for desmoid tumors in familial adenomatous polyposis: an international cohort study. Dis. Colon. Rectum, 54: 1229–1234.
49. Nishida T., Hirota S. (2000) Biological and clinical review of stromal tumors in the gastrointestinal tract. Histol. Histopathol., 15 (4): 1293–1301.
50. O’Brien J.E., Stout A.P. (1964) Malignant Fibrous Xanthomas. Cancer, 17: 1445–1455.
51. Olivier M., Goldgar D.E., Sodha N. et al. (2003) Li-Fraumeni and related syndromes: correlation between tumor type, family structure, and TP53 genotype. Cancer Res., 63: 6643–6650.
52. Ozzello L., Stout A.P., Murray M.R. (1963) Cultural characteristics of malignant histiocytomas and fibrous xanthomas. Cancer, 16: 331–344.
53. Panagopoulos I., Mertens F., Isaksson M. et al. (2001) Clinical impact of molecular and cytogenetic findings in synovial sarcoma. Genes Chromosomes Cancer, 31: 362–372.
54. Perrin R.G., Guha A. (2004) Malignant peripheral nerve sheath tumors. Neurosurg. Clin. N. Am., 15: 203–216.
55. Peter L.M., Lee M.J., Janzen D. et al. (1997) Lipoma and liposarcoma: evaluation using CT and MR imaging. AJR, 169: 589–594.
56. Peterson J., Kransdorf M.J., Bancroft L.W., O’Connor M.I. (2003) Malignant fatty tumors: classification, clinical course, imaging appearance and treatment. Skeletal Radiol., 32: 493–503.
57. Pisters P.W.T., Weiss M., Maki R. (2011) Soft-tissue sarcomas. In: Haller D.G., Wagman L.D., Camphausen C., Hoskins W.J. Cancer Management: A Multidisciplinary Approach Medical, Surgical, & Radiation Oncology (ed 14): UBM Medica LLC.
58. Randall R.L., Albritton K.H., Ferney B.J., Layfield L. (2004) Malignant fibrous histiocytoma of soft tissue: an abandoned diagnosis. Am. J. Orthop. (Belle Mead. NJ), 33(12): 602–608.
59. Sandhu H.S., Sachdeva S., Gill P.K. (2014) Tuberous sclerosis. J. Assoc. Physicians India., 62(7): 602–603.
60. Shukla N.K., Deo S.V.S. (2011) Soft tissue sarcoma-review of experience at a tertiary care cancer centre. Indian J. Surg. Oncol., 2(4): 309–312.
61. Siegel R.L., Miller K.D., Jemal A. (2015) Cancer statistics, 2015. CA A Cancer J., 65: 5–29.
62. Soulié D., Boyer B., Lescop J. et al. (1995) Liposarcome myxoïde. J. Radiol., 76(1): 29–36.
63. Sung M.S., Kang H.S., Lee H.G. et al. (2000) Myxoid liposarcoma: appearance at MR imaging with histologic correlation. Radiol. Graphics., 20: 1007–1019.
64. Suzanne B.K., Kenneth A.J., Nielsen G.P., Andrew E.R. (2001) Orthopadic implant-related sarcoma: a study of twelve cases. Mod. Pathol., 14(10): 969–967.
65. van de Rijn M., Barr F.G., Collins M.H. et al. (1999) Absence of SYT-SSX fusion products in soft tissue tumors other than synovial sarcoma. Am. J. Clin. Pathol., 112: 43–49.
66. Walker D., Gill T.J., Corson J.M. (1971) Leiomyosarcoma in a renal allograft recipient treated with immunosuppressive drugs. JAMA, 215: 2084.
67. Weiss S.W., Goldblum J.R. (2001) Enzinger and Weiss’s soft tissue tumors, 4th Ed. Philadelphia: Mosby-Harcort.
68. Weiss S.W., Goldblum J.R. (2007) St Louis, MO: Mosby, Inc.; Enzinger and Weiss’s Soft Tissue Tumors.
69. Wright P.H., Sim F.H., Soule E.H., Taylor W.F. (1982) Synovial sarcoma. J. Bone Joint Surg., 64(1), 112–122.
70. Zahm S.H., Fraumeni J.F.Jr. (1997) The epidemiology of soft tissue sarcoma. Semin. Oncol., 24: 504–514.
Саркомы мягких тканей: этиология, эпидемиология, классификация
Национальный институт рака, Киев
Резюме. Саркомы мягких тканей — это группа относительно редких злокачественных новообразований различной локализации и гистологической структуры, объединенных общим происхождением из мезодермальных тканей. Развиваясь из эмбриональных мезенхимальных клеток, они впоследствии дифференцируются в клетки поперечнополосатых, гладких мыщц, жировую или фиброзную ткань. По данным различных авторов, саркомы мягких тканей составляют 15% всех злокачественных опухолей у детей и только 1–2,5% — у взрослых. Целью обзора является тщательный анализ существующей литературы по эпидемиологии, этиологии и классификации сарком мягких тканей.
саркома мягких тканей, этиология, эпидемиология, классификация.
Адреса:
Остафійчук Василь Васильович
03022, Київ, вул. Ломоносова, 33/43
Національний інститут раку
Тел.: (044) 257-99-46
E-mail: lugnik2007@gmail.com





Leave a comment