




Грибоподібний мікоз: сучасний стан проблеми в Україні
Крячок І.А.1, Алексик О.М.1, Калмикова А.В.2, Титоренко І.Б.1, Мороз Ю.В.1
- 1Державне некомерційне підприємство «Національний інститут раку», Київ, Україна
- 2Медична лабораторія CSD, Київ, Україна
Резюме. Одним із найбільш актуальних питань є діагностика та лікування шкірних Т-клітинних лімфом (ШТКЛ) з використанням у діагностичній панелі широкого спектра сучасних методів обстеження (гістологічних, імунологічних, молекулярно-генетичних), які дають змогу правильно визначити прогноз та оптимізувати терапію із застосуванням індивідуалізованих підходів до лікування за допомогою новітніх методів. Найпоширенішою формою ШТКЛ є грибоподібний мікоз.
Одержано 11.12.2023
Прийнято до друку 20.12.2023
DOI: 10.32471/clinicaloncology.2663-466X.52-4.31523
ВСТУП
ШТКЛ являють собою гетерогенну групу неходжкінських лімфом, що характеризуються інфільтрацією шкіри пухлинними Т-лімфоцитами. Найпоширенішою формою ШТКЛ є грибоподібний мікоз (mycosis fungoides, ГМ), що становить близько 44% усіх шкірних лімфом [1]. У первинних хворих на ШТКЛ на момент встановлення діагнозу позашкірні прояви переважно відсутні [2].
Річна захворюваність на ШТКЛ становить близько 0,5 на 100 000 осіб, середній вік хворих — від 55 до 60 років. У структурі захворюваності переважають чоловіки (відношення чоловіків до жінок коливається від 2,0:1 до 1,6:1) [3]. Захворюваність на ШТКЛ різко зросла в період з 1973 до 2002 р. і становила близько 3,4% усіх неходжкінських лімфом [4]. До 2013 р. показники захворюваності на ШТКЛ стабілізувалися [5].
Показники захворюваності та виживаності пацієнтів з ГМ, за даними досліджень, зросли з 1969 р. [6–9]. У США захворюваність на ГМ стабілізувалася на рівні 0,41 випадку на 100 000 населення на рік [6, 7, 10, 11]. У даних Департаменту охорони здоров’я та соціального забезпечення Великобританії (Department of Health and Social Care — DHSC) зафіксовано рівень захворюваності на ГМ близько 0,37 випадку на 100 000 осіб на рік [12].
ГМ загалом має сприятливий прогноз із 5-річною виживаністю близько 79–92% [7]. З 1973 р. відмічається позитивна динаміка відносної виживаності [9].
Медіана віку на момент встановлення діагнозу становить близько 55–60 років [13], захворюваність доволі стрімко зростає з віком [14].
Відповідно до результатів ретроспективного дослідження, проведеного у 2020 р., захворюваність підвищилася з 0,3 на 100 000 осіб на рік у 1970-х роках до 0,59 на 100 000 осіб у 2010-х роках. В аналізі виживаності встановлено помітне поліпшення захворювання-специфічної та загальної виживаності з 1970-х до 2010-х років. За даними систематичного огляду медичної літератури, у пацієнтів, у яких патологію діагностовано в період 2000–2016 рр., імовірність встановлення діагнозу ГМ на ранніх (IA–IIA) стадіях захворювання була значно вищою, ніж для пацієнтів, у яких патологію діагностовано до 1997 р. (80% vs 50%, p <0,0001) [15].
У цьому ж дослідженні зафіксовано, що 32–73% пацієнтів, яким встановлено діагноз до 2000 р., мали ранні стадії захворювання, у той час як у період з 2000 до 2016 р. частка осіб, у яких діагностовано патологію на ранніх стадіях, становила 81% (p <0,035 для всіх когорт).
У цих даних встановлено, що підвищення виживаності пацієнтів в останні десятиріччя відбулося хоча б частково завдяки ранній діагностиці ГМ [15].
Імуногістохімічні характеристики гМ
Пухлинні Т-лімфоцити при ГМ переважно мають Т-хелперний імунофенотип: CD4+, CD8-. Співвідношення CD4/CD8-позитивних клітин є одним з діагностичних критеріїв та підкреслює виражене домінування атипових CD4+ лімфоцитів. Часто виявляють аберантну втрату пан-Т-клітинних кластерів диференціювання — CD2, CD3, CD5, CD7, що підкреслює клональну природу пухлинних клітин. Імуногістохімічне дослідження є важливим етапом діагностики ГМ та дозволяє визначити імунофенотип пухлини та провести диференційну діагностику з більш рідкісними Т-клітинними лімфомами. Проточна цитометрія периферичної крові має обмежену роль у діагностиці ГМ, однак є корисною на пізніших стадіях захворювання і може полегшити виявлення клональних Т-клітин, дозволяючи запідозрити ГМ/синдром Сезарі. Для синдрому Сезарі, крім морфологічної оцінки, проточна цитометрія є корисною для виявлення клітин Сезарі, які, як правило, є CD3 та CD4-позитивними та CD8-негативними [16]. Аберантне зникнення антигенів Т-клітин, зокрема CD2, CD3, CD4, CD5, CD7 та CD26, є спільним явищем для синдрому Сезарі та ГМ [17–19]. Одночасна втрата CD7 або CD26 є чутливою та високоспецифічною для синдрому Сезарі [20].
ВТ гМ
Класичними гістологічними ознаками ГМ є епідермотропні малі та середні атипові лімфоцити зі стислими структурами хроматину. Злоякісні лімфоцити більшого розміру з везикулярним ядром є цитологічною ознакою великоклітинної трансформації (ВТ) — одного з негативних прогностичних показників перебігу ГМ [21]. У дослідженнях зафіксовано, що клітини нетрансформованого ГМ та клітини ВТ ГМ того ж пацієнта мають однакове клональне походження [22, 23]. Критерії встановлення ВТ ГМ включають наявність понад 25% злоякісних клітин, діаметр яких у 4 та більше рази перевищує діаметр звичайного малого лімфоцита в інфільтраті [21, 24–26].
ВТ відмічають зі змінною частотою близько у 10–20% випадків на всіх стадіях ГМ; проте найчастіше (близько 50%) виявляють на пізніх стадіях захворювання, наприклад, у хворих з пухлинами шкіри чи ураженням лімфовузлів [27, 28]. Кумулятивна ймовірність трансформації протягом 4 та 12 років захворювання знаходиться на рівні до 21 та 39% відповідно [28].
Наявність ВТ ГМ пов’язана із вищим ризиком рецидиву та прогресії й унаслідок цього з обмеженою специфічною та/або загальною виживаністю [21, 28–35], особливо за наявності клітин ВК у позашкірних ділянках, зокрема в лімфовузлах. Незважаючи на те що явище ВТ у ГМ протягом десятиріч було предметом безлічі описових досліджень, головним чином, спрямованих на клініко-патологічні кореляції, його молекулярний патогенез залишається переважно невідомим і досі. Треба відмітити, що розвиток ВТ у ГМ часто пов’язаний зі збільшеною експресією CD30, білка-рецептора сімейства рецепторів фактора некрозу пухлини (TNFR) на поверхні клітин ГМ. Хоча експресія CD30 не є необхідною умовою встановлення діагнозу ВТ, виникає питання про те, чи може існувати спільний основний молекулярний механізм, який пов’язує обидва цих явища.
У багатовимірному аналізі від 2014 р. виявлено, що вік >60 років та відсутність фіброзу при трансформації незалежно асоціюються із нижчою виживаністю. Проте результати дослідження обмежені недоліками багатовимірної моделі, а саме невеликою кількістю пацієнтів у деяких групах змінних, які, при їх залученні в дослідження, призводили до нестабільних багатовимірних моделей; різниці між типами зразка та розміром біопсій пацієнтів, деякі з яких не завжди містили необхідні для дослідження ознаки, що спричинило відсутність даних та виключення більшості випадків з багатовимірної моделі [36].
Експресія CD30 у гМ
Ефективна системна терапія надзвичайно важлива, особливо у випадках прогресування захворювання. Останнім часом увага приділяється таргетним терапіям, а надійна та стандартизована оцінка біомаркерів набуває важливості. Біомаркери, такі як CD30, гетерогенно експресуються в ГМ (розглянуто в дослідженнях F. Kampa & C. Mitteldorf) [37]. Існують обмежені дані щодо експресії CD30 в ГМ [37–39].
CD30 належить до суперсімейства рецепторів фактора некрозу пухлини. Він викликає тримеризацію TRAF (фактори, асоційовані з рецепторами фактора некрозу пухлини)-зв’язувальних білків (TRAF1, 2 та 5). Це призводить до трансдукції сигналу через MAPK/ERK1/ERK2 (шлях мітоген-активованої протеїнкінази/екстрацелюлярно регульованого шляху протеїнкінази 1, 2) та транскрипції генів за участю NF-κB (ядерний фактор kappa-підсилювач легких ланцюгів активованих В-клітин) [40]. CD30 експресується в різних типах гематологічних захворювань, наприклад, у первинній шкірній анапластичній великоклітинній лімфомі, і має різні біологічні ефекти, які також пов’язані з типом клітин, що експресують CD30, та типом новоутворення [40]. Експресія та функція CD30 також пов’язані з цитокіновим середовищем та складом і функціональним станом мікросередовища пухлини (ТМЕ) [41].
Лікування гМ
Внаслідок гетерогенної та прогресуючої природи ГМ схеми терапії є багатомодальними та змінюються залежно від клінічного етапу, підтипу захворювання та місця ураження [42, 43]. В Європі лікування ГМ, як правило, базується на рекомендаціях консенсусу Європейської організації з дослідження та лікування раку (European Organisation for Research and Treatment of Cancer — EORTC). Зокрема, підходи до лікування хворих з ранніми стадіями ГМ включають очікувальну тактику («Watch and Wait»), топічні кортикостероїди, ретиноїди та інтерферони, низькі дози метотрексату (5–25 мг) та локальну променеву терапію. Серед системних методів на різних стадіях застосовують ретиноїди та інтерферони, УФ-терапію, зокрема PUVA-терапію, яка поєднує в собі використання фотоактивної речовини псоралену (у формі ванн, топічного нанесення чи перорального прийому) з УФ-опроміненням ураженої ділянки шкіри. На пізніх стадіях захворювання використовують системні методи лікування — монохіміотерапію (гемцитабіном, доксорубіцином), поліхіміотерапію за схемою CHOP та подібні їй. Перераховані методи можуть використовуватися в комбінації для досягнення максимального терапевтичного ефекту [44, 45].
Однак досягнення та утримання стійкої відповіді залишається важким завданням, особливо у пацієнтів на пізніх стадіях ГМ. У більшості пацієнтів, які отримують терапію вперше, незалежно від методу лікування в кінцевому підсумку ГМ рецидивує або стає резистентним до лікування [43]. У таких випадках пацієнтам доступні різноманітні нові види терапії, зокрема моноклональними антитілами (могамулізумабом) [46], інгібіторами гістондезацетилази (вориностатом, беліностатом, ромідепсином) [47] та кон’югатами антитіл і препаратів, такими як брентуксимабу ведотин [48, 49].
МАТЕРІАЛИ ТА МЕТОДИ
Робота базується на аналізі випадків хворих на ГМ, діагноз яким встановлено в період з 2012 до 2022 р., за результатами бази даних Національного канцер-реєстру України та бази даних дерматопатологічного відділу діагностичної лабораторії (лабораторної бази), яка є профільною з напрямку онкодерматології за період 2016–2023 рр.
Діагноз ГМ встановлено на основі патогістологічного та імуногістохімічного дослідження вогнищ ураження шкіри. Проведено стадіювання згідно з класифікацією TNMB із використанням даних комп’ютерної томографії шиї, органів грудної, черевної порожнин та малого таза. Враховували показники гемопоезу, радіонуклідних, біохімічних та інших методів обстеження пацієнтів. Обов’язково досліджували показники кістковомозкового кровотворення.
Дослідженням охоплено 177 хворих з Національного канцер-реєстру України (І група) (табл. 1) та 241 пацієнт (ІІ група) з лабораторної бази (табл. 2).
| Стать | Вік, років | Усього | |||||
| 20–29 | 30–39 | 40–49 | 50–59 | 60–69 | понад 70 | ||
| Чоловіки | 1 | 9 | 15 | 22 | 26 | 23 | 96 |
| Жінки | 1 | 8 | 12 | 20 | 22 | 18 | 81 |
| Усього | 2 | 17 | 27 | 42 | 48 | 41 | 177 |
| Стать | Вік, років | Усього | |||||
| 20–29 | 30–39 | 40–49 | 50–59 | 60–69 | понад 70 | ||
| Чоловіки | 9 | 19 | 24 | 34 | 33 | 25 | 144 |
| Жінки | 5 | 12 | 20 | 25 | 18 | 17 | 97 |
| Усього | 14 | 31 | 44 | 59 | 51 | 42 | 241 |
Усі пацієнти отримували спеціальне лікування (імунотерапію, хіміотерапію), деякі з них — фототерапію, променеву терапію на уражені ділянки шкіри.
Більшості хворим проведене імуногістохімічне дослідження, що включало наступну панель CD2, CD3, CD4, CD5, CD7, CD8, CD30, CD56 та Ki-67. Оцінювалося співвідношення CD4:CD8-позитивних клітин, аберантне зникнення пан-Т-клітинних маркерів та частка CD30-позитивних клітин.
РЕЗУЛЬТАТИ
Захворюваність згідно з даними Національного канцер-реєстру України у 2012 р. становила 0,04 на 100 тис. населення, у 2020 р. — 0,07.
Аналізуючи випадки розвитку ГМ, встановлено, що частіше хворіють пацієнти >50 років (74,0% за даними канцер-реєстру та 63,07% випадків за даними лабораторної бази). Захворювання частіше виявляють у чоловіків, ніж у жінок. У результаті аналізу даних І групи показано, що середній вік пацієнтів становив 59±13,56 років (діапазон 29–87), причому 138 пацієнтів (77,9%) мали вік старше 50 років. Чоловіків було 96, жінок 81.
У ІІ групі середній вік пацієнтів становив 54±14,67 років (діапазон 20–82), з яких 152 пацієнти (63,07%) були віком старше 50 років. Чоловіків було 144 особи, жінок — 97. В аналізі 2 баз даних хворих на ГМ виявлено, що розподіл хворих за віком та статтю був подібним.
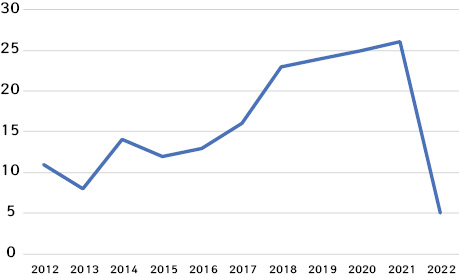
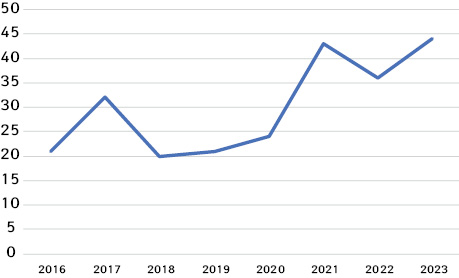
В аналізі кількості випадків за роками зафіксовано, що, починаючи з 2018 р., кількість випадків зростає (рис. 1, 2). Зниження цього показника у 2022 р., вірогідно, пов’язано з воєнними діями в Україні.
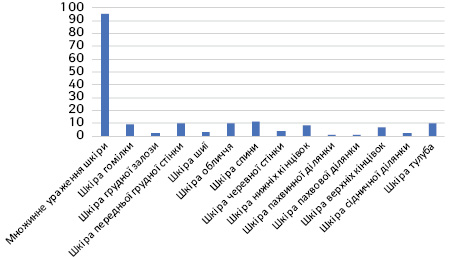
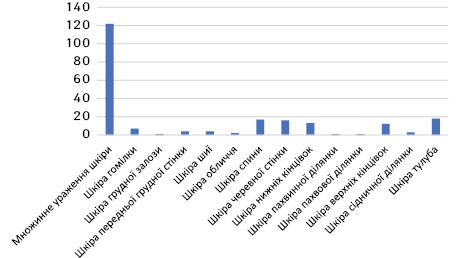
Як виявлено в рис. 3 та 4, в обох базах переважали хворі з множинним ураженням шкіри (54,8 та 50,6% відповідно), що відповідало поширеним стадіям хвороби (ІІІ та ІV стадії).
Під час аналізу рівня експресії СD30 та Ki-67 у 16/62 пацієнтів рівень експресії становив 30% та вище, і всі ці хворі мали ВТ ГМ. Таку ж закономірність відмічали у пацієнтів, які мали експресію Кі-67 на 20% та більше пухлинних клітин (11/62 хворих).
ОБГОВОРЕННЯ
За даними Національного канцер-реєстру України та лабораторної бази, ГМ діагностують частіше у пацієнтів старше 50 років та у чоловіків, ніж у жінок, що не суперечить даним дослідження [3], де середній вік хворих становив від 55 до 60 років та у структурі захворюваності також переважали чоловіки.
При аналізі двох баз також встановлено, що кількість випадків на грибоподібний мікоз в Україні зростає, що можливо пов’язано з покращенням діагностики цього захворювання. Подібна тенденція спостерігається й у світі. Відповідно до результатів ретроспективного дослідження A.E. Kaufman та співавторів, проведеного у 2020 р., рівень захворюваності підвищився з 0,3 на 100 тис. осіб на рік у 1970-х роках до 0,59 на 100 тис. осіб у 2010-х роках [15]. У цьому ж дослідженні встановлено, що 32–73% пацієнтів, яким встановлено діагноз до 2000 р., мали ранні стадії захворювання, водночас як у період з 2000 до 2016 рр. частка пацієнтів, у яких діагностовано патологію на ранніх стадіях, становила 81%. В Україні захворюваність на ГМ залишається доволі низькою: у 2012 р. вона становила 0,04 на 100 тис. населення, у 2020 році — 0,07.
У 62 хворих визначався рівень експресії СD30 та Ki-67 на поверхні пухлинних клітин. У пацієнтів, які мали рівень експресії СD30 на 30% та вище, а також Кі-67 на 20% та вище у пухлинних клітинах діагностовано ВТ ГМ. СD30 вважається одним з негативних прогностичних показників перебігу ГМ [22]. ВТ ГМ виявляють близько у 10–20% випадків на всіх стадіях ГМ; проте найчастіше (приблизно 50%) відмічають на пізніх стадіях захворювання, наприклад, у хворих з пухлинами шкіри чи ураженням лімфовузлів [28, 29]. Згідно з численними дослідженнями [21, 22, 29–35] наявність ВТ ГМ пов’язана із вищим ризиком рецидиву та прогресії захворювання. Також у цих дослідженнях доведено, що розвиток ВТ ГМ часто пов’язаний з підвищеною експресією CD30.
ВИСНОВКИ
Доведено, що хворі з ВТ ГМ мали рівень експресії СD30 на 30% й вище та Кі-67 на 20% та вище пухлинних клітин, що може бути прогностичним маркером негативного прогнозу перебігу захворювання та порушує питання про доцільність подальших досліджень у цьому напрямку.
Визначено, що захворюваність в Україні на ГМ згідно з офіційним показником низька порівняно зі світовими показниками, що, можливо, пов’язано з недоліками статистичного обліку, тривалим періодом спостереження цієї категорії хворих сімейними лікарями, дерматологами без подання даних до Національного канцер-реєстру України та діагностикою захворювання на пізніх стадіях. Останніми роками спостерігається тенденція до збільшення кількості випадків ГМ у реєстрі, що можливо пояснити покращенням діагностичних можливостей в Україні та створенням лабораторії, яка спеціалізується на онкодерматології. Так, стан діагностики та лікування хворих на ГМ в Україні потребує розробки чітких алгоритмів щодо взаємодії лікарів первинної ланки, дерматологів та онкологів, налагодження мультидисциплінарного підходу, покращення статистичного обліку.
СПИСОК ВИКОРИСТАНОЇ ЛІТЕРАТУРИ
1. Lansigan, F., Choi, J., & Foss, F. M. (2008). Cutaneous T-cell lymphoma. Hematology/Oncology Clinics of North America, 22(x), 979–996. doi: 10.1016/j.hoc.2008.07.014.
2. Willemze, R., Jaffe, E. S., Burg, G., Cerroni, L., Berti, E., Swerdlow, S. H., … Meijer, C. J. (2005). WHO-EORTC classification for cutaneous lymphomas. Blood, 105, 3768–3785. doi: 10.1182/blood-2004-09-3502.
3. Rodd, A. L., Ververis, K., & Karagiannis, T. C. (2012). Current and emerging therapeutics for cutaneous T-cell lymphoma: histone deacetylase inhibitors. Lymphoma, 2012, 1–10. doi: 10.1155/2012/290685.
4. Criscione, V. D., & Weinstock, M. A. (2007). Incidence of cutaneous T-cell lymphoma in the United States, 1973–2002. Archives of Dermatological, 143(7), 854–859. doi: 10.1001/archderm.143.7.854.
5. Korgavkar, K., Xiong, M., & Weinstock, M. (2013). Changing incidence trends of cutaneous T-cell lymphoma. JAMA Dermatology, 149(11), 1295–1299. doi: 10.1001/jamadermatol.2013.5526.
6. Bradford, P. T., Devesa, S. S., Anderson, W. F., & Toro, J. R. (2009). Cutaneous lymphoma incidence patterns in the United States: a population-based study of 3884 cases. Blood, 113, 5064–5073. doi: 10.1182/blood-2008-10-184168.
7. Teras, L. R., DeSantis, C. E., Cerhan, J. R., Morton, L. M., Jemal. A., & Flowers, C. R. (2016). 2016 US lymphoid malignancy statistics by World Health Organization subtypes. Cancer Journal for Clinicians, 66, 443–459. doi: 10.3322/caac.21357.
8. Weinstock, M. A., & Gardstein, B. (1999). Twenty-year trends in the reported incidence of mycosis fungoides and associated mortality. American Journal of Public Health, 89, 1240–1244. doi: 10.2105/ajph.89.8.1240.
9. Weinstock, M. A., & Reynes, J. F. (1999). The changing survival of patients with mycosis fungoides. Cancer, 85, 208–212.
10. Korgavkar, K., & Weinstock, M. A. (2014). Changing incidence trends of cutaneous B-cell lymphoma. Journal of Investigative Dermatology, 134, 840–842. doi: 10.1038/jid.2013.393.
11. Criscione, V. D., & Weinstock, M. A. (2007). Incidence of cutaneous T-cell lymphoma in the United States, 1973–2002. Archives of Dermatological, 143, 1973–2002. doi: 10.1001/archderm.143.7.854.
12. Maguire, А., Puelles, J., Raboisson, P., Chavda, R., Gabriel, S., & Thornton, S. (2019). Early-stage mycosis fungoides: epidemiology and prognosis. Acta Dermato-Venereologica, 100(1), adv00013. doi: 10.2340/00015555-3367.
13. Jawed, S. I., Myskowski, P. L., Horwitz, S., Moskowitz, A., & Querfeld, C. (2014). Primary cutaneous T-cell lymphoma (mycosis fungoides and Sezary syndrome): part I. Diagnosis: clinical and histopathologic features and new molecular and biologic markers. Journal of the American Academy of Dermatology, 70, 205.e1–16; quiz 21–2. doi: 10.1016/j.jaad.2013.07.049.
14. Imam, M. H., Shenoy, P. J., Flowers, C. R., Phillips, A., & Lechowicz, M. J. (2013). Incidence and survival patterns of cutaneous T-cell lymphomas in the United States. Leukemia & Lymphoma, 54, 752–759. doi: 10.3109/10428194.2012.729831.
15. Kaufman, A. E., Patel, K., Goyal, K., O’Leary, D., Rubin, N., Pearson, D., … Goyal, A. (2020). Mycosis fungoides: Developments in incidence, treatment and survival. Journal of the European Academy of Dermatology and Venereology, 34, 2288–2294. doi: 10.1111/jdv.16325.
16. Vonderheid, E. C., Bernengo, M. G., Burg, G., Duvic, M., Heald, P., Laroche, L., … Willemze, R. (2002). Update on erythrodermic cutaneous T-cell lymphoma: report of the International Society for Cutaneous Lymphomas. Journal of the American Academy of Dermatology, 46, 95–106. doi: 10.1067/mjd.2002.118538.
17. Hristov, A. C., Vonderheid, E. C., & Borowitz, M. J. (2011). Simplified flow cytometric assessment in mycosis fungoides and Sézary syndrome. American Journal of Clinical Pathology, 136, 944–953. doi: 10.1309/AJCP09OTJOYAVZZK.
18. Jones, D., Dang, N. H., Duvic, M., Washington, L. T., & Huh, Y. O. (2001). Absence of CD26 expression is a useful marker for diagnosis of T-cell lymphoma in peripheral blood. American Journal of Clinical Pathology, 115, 885–892. doi: 10.1309/U1Y6-J4AG-5M4M-7AYV.
19. Ginaldi, L., Matutes, E., Farahat, N., De Martinis, M., Morilla, R., & Catovsky, D. (1996). Differential expression of CD3 and CD7 in T-cell malignancies: a quantitative study by flow cytometry. British Journal of Haematology, 93, 921–927. doi: 10.1046/j.1365-2141.1996.d01-1720.x.
20. Boonk, S. E., Zoutman, W. H., Marie-Cardine, A., van der Fits, L., Out-Luiting, J. J., Mitchell, T. J., … Vermeer, M. H. (2016). Evaluation of immunophenotypic and molecular biomarkers for Sézary syndrome using standard operating procedures: a multicenter study of 59 patients. Journal of Investigative Dermatology, 136, 1364–1372. doi: 10.1016/j.jid.2016.01.038.
21. Salhany, K. E., Cousar, J. B., Greer, J. P., Casey, T. T., Fields, J. P., & Collins, R. D. (1988). Transformation of cutaneous T cell lymphoma to large cell lymphoma. A clinicopathologic and immunologic study. American Journal of Pathology, 132, 265–277.
22. Wolfe, J. T., Chooback, L., Finn, D. T., Jaworsky, C., Rook, A. H., & Lessin, S. R. (1995). Large-cell transformation following detection of minimal residual disease in cutaneous T-cell lymphoma: Molecular and in situ analysis of a single neoplastic T-cell clone expressing the identical T-cell receptor. Journal of Clinical Oncology, 13, 1751–1757. doi: 10.1200/JCO.1995.13.7.1751.
23. Wood, G. S., Bahler, D. W., Hoppe, R. T., Warnke, R. A., Sklar, J. L., & Levy, R. (1993). Transformation of mycosis fungoides: T-cell receptor beta gene analysis demonstrates a common clonal origin for plaque-type mycosis fungoides and CD30+ large-cell lymphoma. Journal of Investigative Dermatology, 101, 296–300. doi: 10.1111/1523-1747.ep12365416.
24. Herrmann, J. L., & Hughey, L. C. (2012). Recognizing large-cell transformation of mycosis fungoides. Journal of the American Academy of Dermatology, 67, 665–672. doi: 10.1016/j.jaad.2011.12.011.
25. Stefanato, C. M., Tallini, G., & Crotty, P. L. (1998). Histologic and immunophenotypic features prior to transformation in patients with transformed cutaneous T-cell lymphoma: Is CD25 expression in skin biopsy samples predictive of large cell transformation in cutaneous T-cell lymphoma? American Journal of Clinical Dermatology, 20, 1–6. doi: 10.1097/00000372-199802000-00001.
26. Cerroni, L., Rieger, E., Hödl, S., & Kerl, H. (1992). Clinicopathologic and immunologic features associated with transformation of mycosis fungoides to large-cell lymphoma. American Journal of Surgical Pathology, 16, 543–552. doi: 10.1097/00000478-199206000-00002.
27. Barberio, E., Thomas, L., Skowron, F., Balme, B., & Dalle, S. (2007). Transformed mycosis fungoides: Clinicopathological features and outcome. British Journal of Dermatology, 157, 284–289. doi: 10.1111/j.1365-2133.2007.08008.x.
28. Diamandidou, E., Colome-Grimmer, M., Fayad, L., Duvic, M., & Kurzrock, R. (1998). Transformation of Mycosis Fungoides/Sezary Syndrome: Clinical Characteristics and Prognosis. Blood, 92, 1150–1159. doi: 10.1182/blood.V92.4.1150.
29. Scarisbrick, J. J. (2017). Prognostic factors in mycosis fungoides: International advances in the validation of prognostic indices. British Journal of Haematology, 176, 1129–1130. doi: 10.1111/bjd.15214.
30. Li, G., Chooback, L., Wolfe, J. T., Rook, A. H., Felix, C. A., Lessin, S. R., & Salhany, K. E. (1998). Overexpression of p53 protein in cutaneous T cell lymphoma: Relationship to large cell transformation and disease progression. Journal of Investigative Dermatology, 110, 767–770. doi: 10.1046/j.1523-1747.1998.00167.x.
31. Arulogun, S. O., Prince, H. M., Ng, J., Lade, S., Ryan, G. F., Blewitt, O., & McCormack, C. (2008). Long-term outcomes of patients with advanced-stage cutaneous T-cell lymphoma and large cell transformation. Blood, 112, 3082–3087. doi: 10.1182/blood-2008-05-154609.
32. Vergier, B., de Muret, A., Beylot-Barry, M., Vaillant, L., Ekouevi, D., Chene, G., … Wechsler, J. (2000). Transformation of mycosis fungoides: Clinicopathological and prognostic features of 45 cases. French Study Group of Cutaneious Lymphomas. Blood, 95, 2212–2218.
33. Benner, M. F., Jansen, P. M., Vermeer, M. H., & Willemze, R. (2012). Prognostic factors in transformed mycosis fungoides: A retrospective analysis of 100 cases. Blood, 119, 1643–1649. doi: 10.1182/blood-2011-08-376319.
34. Greer, J. P., Salhany, K. E., Cousar, J. B., Fields, J. P., King, L. E., Graber, S. E., … Collins, R. D. (1990). Clinical features associated with transformation of cerebriform T-cell lymphoma to a large cell process. Hematological Oncology, 8, 215–227. doi: 10.1002/hon.2900080406.
35. Agar, N. S., Wedgeworth, E., Crichton, S., Mitchell, T. J., Cox, M., Ferreira, S., … Whittaker, S.J. (2010). Survival outcomes and prognostic factors in mycosis fungoides/Sézary syndrome: Validation of the revised International Society for Cutaneous Lymphomas/European Organisation for Research and Treatment of Cancer staging proposal. Journal of Clinical Oncology, 28, 4730–4739. doi: 10.1200/JCO.2009.27.7665.
36. Pulitzer, M., Myskowski, P. L., Horwitz, S. M., Querfeld, C., Connolly, B., Li, J., & Murali, R. (2014). Mycosis fungoides with large cell transformation: Clinicopathological features and prognostic factors. Pathology, 46, 610–616. doi: 10.1097/PAT.0000000000000166.
37. Kampa, F., & Mitteldorf, C. (2021). A review of CD30 expression in cutaneous neoplasms. Journal of Cutaneous Pathology, 48(4), 495–510. doi: 10.1111/cup.13894.
38. Koens, L., van de Ven, P. M., Hijmering, N. J., Kersten, M. J., Diepstra, A., Chamuleau, M., & de Jong, D. (2018). Interobserver variation in CD30 immunohistochemistry interpretation; consequences for patient selection for targeted treatment. Histopathology, 73, 473–482. doi: 10.1111/his.13647.
39. Xu, M. L., Gabali, A., Hsi, E. D., Fedoriw, Y., Vij, K., Salama, M. E., … Gru, A. A. (2020). Practical approaches on CD30 detection and reporting in lymphoma diagnosis. American Journal of Surgical Pathology, 44, e1–e14. doi: 10.1097/PAS.0000000000001368.
40. Horie, R., & Watanabe, T. (1998). CD30: Expression and function in health and disease. Seminars in Immunology, 10, 457–470. doi: 10.1006/smim.1998.0156.
41. Muta, H., & Podack, E. R. (2013). CD30: From basic research to cancer therapy. Immunologic Research, 57, 151–158. doi: 10.1007/s12026-013-8464-1.
42. Jonak, C., Tittes, J., Brunner, P. M., & Guenova, E. (2021). Mycosis fungoides and Sézary syndrome. Journal der Deutschen Dermatologischen Ges, 19, 1307–1334. doi: 10.1016/j.hoc.2018.09.001.
43. Kamijo, H., & Miyagaki, T. (2021). Mycosis fungoides and Sézary syndrome: Updates and review of current therapy. Current Treatment Options in Oncology, 22, 10. doi: 10.1007/s11864-020-00809-w.
44. Trautinger, F., Knobler, R., Willemze, R., Peris, K., Stadler, R., Laroche, L., … Trautinger, F. (2006). EORTC consensus recommendations for the treatment of mycosis fungoides/Sézary syndrome. European Journal of Cancer, 42, 1014–1030. doi: 10.1016/j.ejca.2023.113343.
45. Latzka, J., Assaf, C., Bagot, M., Cozzio, A., Dummer, R., Guenova, E., … Knobler, R. (2023). European Organisation for Research and Treatment of Cancer consensus recommendations for the treatment of mycosis fungoides/Sézary syndrome—Update 2023. European Journal of Cancer, 195, 113343. doi: 10.1016/j.ejca.2017.02.027.
46. Duvic, M., Pinter-Brown, L. C., Foss, F. M., Sokol, L., Jorgensen, J. L., Challagundla, P., … Kim, Y. H. (2015). Phase 1/2 study of mogamulizumab, a defucosylated anti-CCR4 antibody, in previously treated patients with cutaneous T-cell lymphoma. Blood, 125, 1883–1889. doi: 10.1182/blood-2014-09-600924.
47. Yoon, S., & Eom, G. H. (2016). HDAC and HDAC inhibitor: From cancer to cardiovascular diseases. Chonnam Medical Journal, 52, 1–11. doi: 10.4068/cmj.2016.52.1.1.
48. Kim, Y. H., Tavallaee, M., Sundram, U., Salva, K. A., Wood, G. S., Li, S., … Horwitz, S. M. (2015). Phase II investigator-initiated study of brentuximab vedotin in mycosis fungoides and Sézary syndrome with variable CD30 expression level: A multi-institution collaborative project. Journal of Clinical Oncology, 33, 3750–3758. doi: 10.1200/JCO.2014.60.3969.
49. Assaf, C., Waser, N., Bagot, M., He, M., Li, T., Dalal, M., … Illidge, T. M. (2021). Contemporary treatment patterns and response in relapsed/refractory cutaneous T-cell lymphoma (CTCL) across five European countries. Cancers, 14, 145. doi: 10.3390/cancers14010145.
Адреса для листування:
Алексик Олена Михайлівна
03022, Київ, вул. Здановської Юлії, 33/43
Державне некомерційне підприємство «Національний інститут раку»
E-mail: aleksikel@gmail.com
Correspondence:
Olena Aleksyk
33/43 Yulia Zdanovskaya str., Kyiv, 03022
Nonprofit Organization National Cancer Institute
E-mail: aleksikel@gmail.com





Leave a comment