




Терапевтичні підходи до лікування дорослих пацієнтів з гістіоцитозом із клітин Лангерганса
Крячок И.А., Скрипец Т.В., Новосад И.О., Новдах О.П.
Резюме. Гістіоцитоз із клітин Лангерганса — це орфанне захворювання, в основі якого лежить клональна проліферація патологічних гістіоцитів, які фенотипічно схожі з клітинами Лангерганса, що формують специфічні інфільтрати в різних органах і тканинах. У статті запропоновано рекомендації, прийняті міжнародною групою з вивчення проблем діагностики та лікування гістіоцитозу з клітин Лангерганса. Градація рекомендацій, наведена в цій статті, заснована на даних доказової медицини та угодах між експертами.
Резюме. Гистиоцитоз из клеток Лангерганса — орфанное заболевание, в основе которого лежит клональная пролиферация патологических гистиоцитов, фенотипически схожих с клетками Лангерганса, которые формируют специфические инфильтраты в различных органах и тканях. В данной статье представлены рекомендации, принятые международной группой по изучению проблемы диагностики и лечения гистиоцитоза из клеток Лангерганса. Градация рекомендаций, приведенная в статье, основана на данных доказательной медицины и соглашениях между экспертами.

Общие аспекты
Гистиоцитоз из клеток Лангерганса (ГКЛ) — редкое заболевание, в основе которого лежит клональная пролиферация патологических гистиоцитов, фенотипически схожих с клетками Лангерганса, которые формируют специфические инфильтраты в различных органах и тканях.
Согласно современным подходам, ГКЛ — заболевание иммунной системы с патологическим иммунным ответом, приводящим к пролиферации клеток Лангерганса, эозинофильной инфильтрации, образованию гранулем, фиброза, остеолитических очагов и т.д. [1].
Заболеваемость у детей составляет 3–5 случаев на 1 млн населения в год, у взрослых — 1–2 случая на 1 млн жителей. Мужчины болеют чаще, чем женщины.
Этиология и патогенез ГКЛ остаются неизвестными. Канцерассоциированная мутация (BRAF V600E) определяется в более чем половине исследованных образцов, течение заболевания имеет хронический характер с чередованиями рецидивов и ремиссий.
ГКЛ может поражать любой орган или систему органов. Наиболее часто поражению подлежат кости, кожа и гипофиз, реже — лимфатические узлы, печень, селезенка, кишечник, центральная нервная система (ЦНС), кроветворная система. Легкие могут вовлекаться в патологический процесс одновременно с другими органами или последовательно, но изолированный легочный ГКЛ часто возникает у взрослых и может затем трансформироваться в мультисистемное поражение.
Диагноз
Диагноз ГКЛ основан на гистологическом и иммунофенотипическом исследовании пораженных тканей.
Клетки Лангерганса имеют плотную эозинофильную цитоплазму, хотя в отдельных клетках цитоплазма может быть пенистой, вакуолизированной, содержать фагоцитированные включения и гемосидерин. Ядра клеток крупные, бледные, с нежным хроматином и мелкими ядрышками, контуры их изрезанные, форма овальная, расщепленная или бобовидная. Ультраструктурно в этих клетках выявляют гранулы Бирбека, которые напоминают застежку-молнию, что является специфическим признаком ГКЛ (так называемый золотой стандарт). Выявляемые клетки Лангерганса экспрессируют антигены CD45+, S100+, CD1a+, CD14– на поверхности мембран [2] (рис. 1, 2).
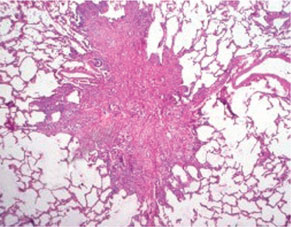
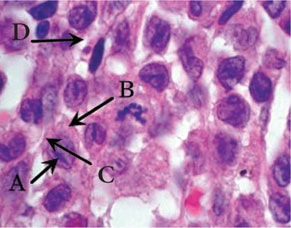
Клиническая картина
Наиболее распространенными симптомами ГКЛ являются одышка, кашель, боль в костях, образование мягкотканных компонентов, сыпь, зуд, жажда и лимфаденопатия [3]. Пациенты отмечают усталость, общую слабость, уменьшение массы тела, ночное повышенное потоотделение, тошноту и лихорадку. Классическая триада ГКЛ включает дефекты черепных костей, экзофтальм, несахарный диабет. Несахарный диабет развивается у 30% пациентов c ГКЛ [1], но может достигать и 40% у пациентов с мультисистемной формой ГКЛ или 94% — в случае гипофизарной недостаточности [4, 5].
Диагностика
Лабораторная диагностика ГКЛ должна быть проведена всем без исключения пациентам, вне зависимости от количества пораженных органов или систем органов (табл. 1). Рекомендуемое обследование для установления диагноза и оценки распространенности заболевания включает: общий анализ крови, биохимический анализ крови, коагулограмму, общий анализ мочи, ультразвуковое исследование (УЗИ) органов брюшной полости, определение уровня тиреотропного гормона (ТТГ) и свободного тироксина Т4.
Таблица 1. Лабораторные исследования, клинические данные для специфического клинического исследования, а также определения «органов риска»
|
Показания |
Исследование |
| Бицитопения, панцитопения или персистирующая необъяснимая цитопения | Аспирация костного мозга или трепанобиопсия для исключения иных заболеваний |
| Печеночная дисфункция | Биопсия печени рекомендуется только в случаях клинических признаков вовлечения органа. От результата биопсии будет зависеть дальнейшая тактика лечения (дифференциальная диагностика между ГКЛ и склерозирующим холангитом) |
| Поражение легких (изменения на рентгенограмме или симптомы/признаки вовлечения легких) | КТ органов грудной полости высокого разрешения спирометрия |
| Подозрения на поражение костей черепа, включая верхнюю и нижнюю челюстные кости | Магнитно-резонансная томография (МРТ) черепа |
| Поражение позвоночника | МРТ позвоночника (для исключения компрессии спинного мозга) |
| Визуальные или неврологические изменения | МРТ головы Неврологическое обследование Нейропсихологическое исследование |
| Нарушение слуха, поражение височной и решетчатой кости | МРТ головы Консультация оториноларинголога КТ височной/решетчатой кости |
| Необъяснимая диарея, мальабсорбция | Эндоскопическое исследование Биопсия |
Первоочередными рентгенологическими методами исследования должны быть рентгенография органов грудной полости (прямая и боковая проекции), а также компьютерная томография (КТ) костей скелета и черепа [6].
Характерным симптомокомплексом при ГКЛ является триада Крисчена: дефекты развития черепа, экзофтальм и несахарный диабет [7].
Стратификация
1. Мультисистемная форма ГКЛ (Multisystem Langerhans cell histiocytosis — MS-LCH): с/без вовлечения «органов риска».
2. Локализованная форма ГКЛ (Single system Langerhans cell histiocytosis — SS-LCH): очаговое или мультифокальные поражения.
3. Локализованная форма ГКЛ (SS-LCH): вовлечение «особых зон».
Определение вовлеченных органов
После установления диагноза ГКЛ должны быть оценены и определены вовлеченные органы в соответствии с клиническими, биологическими и радиологическими критериями.
Существуют так называемые органы риска — это печень, селезенка, костный мозг, ЦНС. Вовлечение «органов риска» свидетельствует о неблагоприятном прогнозе течения заболевания.
Кроме «органов риска», выделяют «особые зоны», которые могут привести к быстрому развитию поражения ЦНС из-за критичного анатомического строения. Вовлечение костей черепа на протяжении длительного времени обусловливает развитие несахарного диабета. В исследованиях DALHX 83 и DALHX 90 у пациентов с мультисистемной формой ГКЛ и вовлечением лицевого черепа, в частности с поражением зрительного и слухового аппарата, ротовой полости, на момент установления диагноза повышается риск развития несахарного диабета [8]. При сравнении MS-LCH и SS-LCH авторы выявили, что поражения слухового, зрительного аппарата, ротовой полости и комбинированные черепно-лицевые повреждения являются статистически значимыми, и риск поражения ЦНС повышается, если заболевание остается активным в течение длительного периода или прогрессирует.
Следует отметить, что поражение определенного очага, например позвонков с вовлечением окружающих мягких тканей, выделяется как «особая зона», поскольку такие очаги могут привести к незамедлительной прогрессии заболевания.
Все «особые зоны» представлены ниже [9]:
- поражение костей лицевого черепа (орбитальная, височная, клиновидная, скуловая или решетчатая кость; верхняя челюсть, околоносовые пазухи) или черепной ямки с внутричерепным мягкотканным компонентом;
- вовлечение зрительного аппарата (экзофтальм или поражение орбитальной, скуловой или клиновидной кости);
- вовлечение слухового аппарата (проявляется наружным отитом, средним отитом, отореей или повреждением в височной кости, сосцевидном отростке височной кости или каменистой части височной кости);
- вовлечение ротовой полости (поражение слизистой оболочки полости рта, десен, небных костей, верхней и нижней челюсти).
Следует помнить, что поражения позвонков без вовлечения окружающих мягких тканей не выделяется как «особая зона». А если при очаговой форме заболевания поражена одна из «особых зон», это свидетельствует о необходимости назначения системной терапии.
Оценка факторов риска
При вовлечении в процесс кроветворной системы развивается анемия, лейкоцитопения, тромбоцитопения, увеличение селезенки >2 см ниже реберной дуги по среднеключичной линии. При поражении печени ее размеры увеличиваются >3 см ниже реберной дуги по среднеключичной линии и/или наблюдается гипопротеинемия <55 г/л, гипоальбуминемия <25 г/л. При вовлечении в процесс легких возникают изменения на КТ. Рекомендуется проводить диагностику бронхоальвеолярного лаважа (диагностическая бронхоскопия): в лаважной жидкости при этом выявляют >5% CD1a+ клеток. Если результаты бронхоальвеолярного лаважа не информативны и CD1a не определяется, проводится биопсия легкого [10].
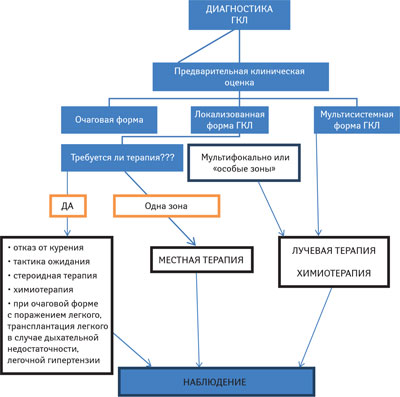
Алгоритм терапии при ГКЛ
При некоторых локализациях, вне зависимости от того, есть ли у больного очаговое поражение или локализованная форма, следует рассмотреть назначение системной терапии:
- локализованная форма ГКЛ с риском поражения ЦНС;
- локализованная форма ГКЛ с мультифокальным поражением костей;
- локализованная форма ГКЛ с поражением «особых зон».
Наблюдение, местная или «не интенсивная системная» терапия
Выбор тактики терапии зависит от локализации, размеров очага поражения, симптомов заболевания. В случае локализованной формы ГКЛ с однофокальным поражением костей без риска развития поражения ЦНС рекомендуется местная терапия и постоянное наблюдение (рис. 3). Хирургическое удаление очагов поражения в костях не рекомендуется, так как это может привести к увеличению размеров дефектов и времени заживления. Для скорейшего заживления можно применить введение метилпреднизолона в дозе 40–160 мг прямо в очаг поражения в кости. При множественном поражении костей при ГКЛ следует применять системную терапию. При ограниченных поражениях кожи серьезные хирургические вмешательства не применяют. Нанесения 20% азотистого иприта на пораженный участок кожи является эффективным методом лечения у детей, но данные о применении его у взрослых ограничены [11]. Псорален сочетанно с ультрафиолетовым излучением (ПУВА-терапия) является эффективной опцией для лечения кожных проявлений при ГКЛ [12]. Однако если поражения находятся в области кожных складок, подмышечных впадинах, паховых, меж- и инфрамаммарных регионах, на голове, доступ к ним осуществить этим методом достаточно трудно. Противопоказанием к ПУВА-терапии является зона мужских половых органов [13].
Применение талидомида (в дозе 100 мг/сут) при кожных поражениях ГКЛ достаточно эффективно для пациентов, у которых нет вовлечения в процесс печени, селезенки и костного мозга [14]. Однако при изолированной терапии талидомидом высока вероятность развития нейротоксичности [15].
У пациентов с кожными проявлениями при ГКЛ возможно назначение азатиоприна (метаболит 6-меркаптопурина (6-МП)), но отсутствие достаточной доказательной базы является препятствием для внесения данного препарата в стандарты лечения при этой патологии. При назначении азатиоприна пациенту следует сделать анализ на тиопурин S-метилтрансферазу (ТПМТ). ТПМТ — ключевой фермент катаболизма меркаптопурина (6-МП). Мутации в гене ТПМТ приводят к снижению активности фермента и развитию тяжелой гематологической токсичности при приеме 6-МП в стандартных дозах (50—75 мг/м/сут). Если активность фермента снижена, то при введении стандартных доз 6-МП образуется большое количество тиогуаниновых нуклеотидов, что в большинстве случаев приводит к возникновению тяжелых побочных эффектов, например миелосупрессии. Видимые изменения наблюдаются через 6 нед приема азатиоприна.
Существуют опубликованные данные об успешном применении низких доз метотрексата (20 мг 1 раз в неделю) в монотерапии или в сочетании с азатиоприном или преднизолоном [16].
Системная терапия
В качестве терапии первой линии для всех больных с мультисистемным гистиоцитозом рекомендуется 6-недельный курс с применением винбластина и преднизолона (рис. 4). Выбор лечения не зависит от вовлечения «органов риска». Дальнейшая терапия обусловливается ответом пациента на первичную терапию. Для того чтобы определить ответ на терапию, необходима оценка лечения в конце первого 6-недельного курса [9].
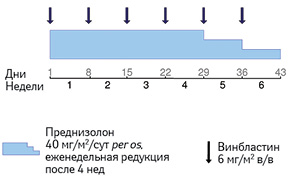
Адаптировано из [9]
Для пациентов без вовлечения «органов риска», которые не показали положительной динамики, и для пациентов с вовлечением «органов риска», которые ответили на первый курс лечения, рекомендуется проведение второго курса терапии с применением винбластина и преднизолона (рис. 5).
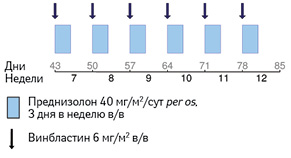
Адаптировано из [9]
Кроме того, Европейская Ассоциация по разработке стандартов терапии гистиоцитоза (Histiocyte Society — HS) рекомендует всем пациентам, которые достигли полного ответа через 6–12 нед инициальной терапии, продолжить поддерживающее лечение. Поддерживающая терапия состоит из введения винбластина и преднизолона каждые 3 нед и ежедневного непрерывного приема 6-МП, при этом общий период лечения составляет до 12 мес (рис. 6).
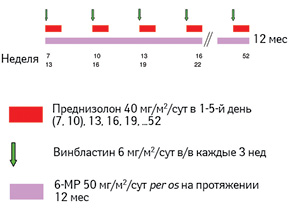
Адаптировано из [9]
В связи с высоким уровнем нейротоксичности и часто развивающимися побочными эффектами, индуцированными стероидами, некоторые эксперты предпочитают монотерапию кладрибином, цитарабином или этопозидом [17].
В ретроспективном исследовании, проведенном M.A. Cantu и соавторами, оценивали результаты лечения 58 пациентов с поражениями костей. Авторы отметили явное преимущество использования цитарабина по сравнению со схемами винбластин/преднизолон с точки зрения ответа на терапию и токсичности [18]. Интенсивные схемы химиотерапии (например MACOP-B) эффективны, однако их применение оправдано только при агрессивном течении ГКЛ.
В последнее время, в случае вовлечения «органов риска» или при опухолевом поражении головного мозга, многообещающей является комбинация 2-хлордезоксиаденозина (2-CdA) и цитарабина. Однако необходимо принять во внимание, что в исследование F. Bernard и соавторов [19] были включены дети, а не взрослые.
Оценка ответа
Оценку ответа проводят после 2–3 курсов химиотерапии при рестадировании. Должна быть применена полная лабораторная и рентгенологическая диагностика, выполняемая перед началом терапии.
Сальвадж-терапия
Пациенты с вовлечением «органов риска» на момент установления диагноза и те, кто не продемонстрировал положительной динамики после терапии первой линии, являются кандидатами для проведения сальвадж-терапии (терапии спасения). Терапия спасения также рекомендуется для пациентов, у которых после проводимого лечения первой линии развилось поражение «органов риска».
Терапию рефрактерных форм заболевания следует проводить с использованием препаратов, которые не применялись в первой линии терапии. В случае прогрессии заболевания, особенно при вовлечении ЦНС, к 2-CdA может быть добавлен цитарабин (оба препарата проникают через гематоэнцефалический барьер) [20]. Существуют опубликованные данные о применении ингибиторов тирозинкиназы (иматиниб) [21, 22]. В редких случаях агрессивного течения заболевания успешно применяется трансплантация стволовых клеток периферической крови [23, 24].
В настоящее время отсутствует достаточная доказательная база относительно выбора оптимального курса терапии для пациентов с прогрессией заболевания.
Результаты, актуальные на данный момент, основаны на ограниченном количестве наблюдений и должны быть подтверждены проспективными клиническими исследованиями. Тем не менее комбинированная схема с использованием 2-CdA (кладрибин) и цитарабина (Ара-С), а также трансплантация стволовых клеток после режима кондиционирования сниженной интенсивности (РИК-SCT) демонстрируют удовлетворительные результаты. Европейская Ассоциация по разработке стандартов терапии гистиоцитоза в настоящее время проводит исследования по двум вышеупомянутым протоколам.
Лучевая терапия
Лучевая терапия проводится в случае неврологических нарушений и высокой возможности хирургического вмешательства, например при поражении альвеолярных отростков верхней и нижней челюсти или основания черепа [25]. Рекомендации касательно доз облучения до сих пор обсуждаются (табл. 2). Для взрослых возможно назначение лучевой терапии в суммарной очаговой дозе 10–20 Гр, однако в некоторых случаях можно применять и в дозе до 45 Гр. Лучевая терапия является эффективной опцией лечения в определенных ситуациях [26, 27]. Ремиссии регистрируют у 79–100% пациентов.
Таблица 2. Возможные показания для применения лучевой терапии у взрослых
|
Рекомендации |
|
Изолированное «неоперабельное» поражение:
|
|
Рефрактерное течение или прогрессия заболевания:
|
|
Адъювантная терапия в случае краевой или неполной резекции:
|
ЦНС
Лечение пациентов с поражением ЦНС требует индивидуального подхода и зависит от формы и стадии заболевания, а также от предварительно полученного лечения и, таким образом, должно быть определено индивидуально.
Ответ на первоначальную терапию является важным прогностическим фактором. Для пациентов с мультисистемным гистиоцитозом без вовлечения «органов риска» вероятность выживаемости составляет около 95% при лечении стандартными режимами с винбластином и стероидами. Результаты международных исследований последних 20 лет доказывают, что комбинация винбластина с преднизолоном обеспечивает успех в лечении у пациентов при поражении ЦНС, сходный с таковым у больных с мультисистемным гистиоцитозом. Данные последнего проспективного исследования LCH III подтверждают, что этот режим терапии наиболее приемлем при мультисистемной форме гистиоцитоза с и без вовлечения «органов риска».
В настоящее время нет убедительной доказательной базы относительно оптимального курса лечения у группы пациентов с MS-LCH [9].
Наблюдение
ГКЛ может рецидивировать в любой момент, а также привести к дисфункции органов и систем. Поэтому необходимо проводить наблюдение пациентов, а также мониторинг функциональных нарушений (табл. 3).
Таблица 3. Наблюдение пациентов
|
Клиническое обследование |
1-й год |
2–5-й годы |
|
Каждые 6 нед |
Каждые 6 мес |
|
|
Лабораторные тесты для исследования функций пораженного органа: общий анализ крови, скорость оседания эритроцитов, печеночные и почечные тесты, осмолярность мочи |
Каждые 3 мес |
Ежегодно |
|
Рентгенография пораженных костей |
Только если есть новые поражения или возможен рецидив |
Только если есть новые поражения или возможен рецидив |
|
Аудиограмма для пациентов с вовлечением слухового аппарата/сосцевидного отростка |
В течение 1 года |
В течение 5 лет |
|
КТ органов грудной полости у пациентов с поражением легких |
Каждые 6 мес |
Только при подозрении на прогрессию |
|
УЗИ у пациентов с поражением печени |
Каждые 6 мес |
Ежегодно |
|
МРТ черепа у пациентов с несахарным диабетом или другими эндокринопатиями или пациентов с риском развития поражения ЦНС |
1 раз в год |
Каждые 2 года |
|
Нейропсихометрическая оценка пациентов с поражением ЦНС |
1 раз в год |
Каждые 2 года |
Выводы
На сегодня унифицированных стандартов диагностики и лечения взрослых пациентов с ГКЛ не существует, так как все рекомендации базируются на исследованиях в педиатрии. Взрослые больные могут получать терапию по протоколам, разработанным для пациентов детского возраста. Однако данная терапия не всегда эффективна и более токсична. Ныне необходимо проведение дальнейших исследований для усовершенствования и написания стандартов по тактике ведения взрослых пациентов с ГКЛ.
Список использованной литературы
1. Chikwava K., Jaffe R. (2004) Langerin (CD207) staining in normal pediatric tissues, reactive lymph nodes, and childhood histiocytic disorders. Pediatr. Dev. Pathol., 7(6): 607–614.
2. Jaffe R., Weiss L.M., Facchetti F. (2008) Tumors derived from Langerhans cells. In: Swerdlow S.H., Campo E., Harris N.L. et al., eds. WHO Classification of Tumors of hematopoietic and lymphoid tissues. Lyon: IARC Pressp: 358–360.
3. Lee J.S., Ko G.H., Kim H.C. et al. (2006) Langerhans cell sarcoma arising from Langerhans cell histiocytosis: a case report. J. Korean. Med. Sci., 21(3): 577–580.
4. Kaltsas G.A., Powles T.B., Evanson J. et al. (2000) Hypothalamo-pituitary abnormalities in adult patients with Langerhans cell histiocytosis: clinical, endocrinological, and radiological features and response to treatment. J. Clin. Endocrinol. Metab., 85(4): 1370–1376.
5. Makras P., Alexandraki K.I., Chrousos G.P. et al. (2007) Endocrine manifestations in Langerhans cell histiocytosis. Trends Endocrinol. Metab., 18(6): 252–257.
6. Tazi A. (2006) Adult pulmonary Langerhans’ cell histiocytosis. Eur. Respir. J., 27(6): 1272–1285.
7. Alexandraki K.I., Makras P., Protogerou A.D. et al. (2008) Cardiovascular risk factors in adult patients with multisystem Langerhans-cell histiocytosis: evidence of glucose metabolism abnormalities. QJM, 101(1): 31–40.
8. Grois N., Potschger U., Prosch H. et al. (2006) Risk factors for diabetes insipidus in langerhans cell histiocytosis. Pediatr. Blood Cancer, 46(2): 228–233.
9. Minkov M., Grois N., McClain K., Nanduri V., Rodriguez-Galindo C., Simonitsch-Klupp I., Visser J., Weitzman Sh., Whitlock J., Windebank K. (2009) Histiocyte Society. Evaluation and Treatment Guidelines, April 2009, 21 p.
10. Girschikofsky M., Arico M., Castillo D. et al. (2013) Management of adult patients with Langerhans cell histiocytosis: recommendations from an expert panel on behalf of Euro-Histio-Net. Orphanet J. Rare Dis., 8(Article 72): 5 (http://www.ojrd.com/ content/8/1/72).
11. Hoeger P.H., Nanduri V.R., Harper J.I. et al. (2000) Long term follow up of topical mustine treatment for cutaneous Langerhans cell histiocytosis. Arch. Dis. Child, 82(6): 483–487.
12. Sakai H., Ibe M., Takahashi H. et al. (1996) Satisfactory remission achieved by PUVA therapy in Langerhans cell histiocytosis in an elderly patient. J. Dermatol., 23(1): 42–46.
13. Imafuku S., Shibata S., Tashiro A., Furue M. (2007) Cutaneous Langerhans cell histiocytosis in an elderly man successfully treated with narrowband ultraviolet B. Br. J. Dermatol., 157(6): 1277–1279.
14. Sander C.S., Kaatz M., Elsner P. (2004) Successful treatment of cutaneous langerhans cell histiocytosis with thalidomide. Dermatology, 208(2): 149–152.
15. McClain K.L., Kozinetz C.A. (2007) A phase II trial using thalidomide for Langerhans cell histiocytosis. Pediatr. Blood Cancer, 48(1): 44–49.
16. Steen A.E., Steen K.H., Bauer R., Bieber T. (2001) Successful treatment of cutaneous Langerhans cell histiocytosis with low-dose methotrexate. Br. J. Dermatol., 145(1): 137–140.
17. Chu A. (2011) Dermatological Aspects and Presentation of an Adult Clinic. Oral Presentation at the Annual Meeting of the Histiocyte Society, Vienna, 2011.
18. Cantu M.A. (2012) Optimal therapy for adults with Langerhans cell histiocytosis bone lesions. PloSOne, 7(8): e43257.
19. Bernard F., Thomas C., Bertrand Y. et al. (2005) Multi-centre pilot study of 2-chlorodeoxyadenosine and cytosine arabinoside combined chemotherapy in refractory Langerhans cell histiocytosis with haematological dysfunction. Eur. J. Cancer, 41(17): 2682–2689.
20. McClain K.L. (2005) Drug therapy for the treatment of Langerhans cell histiocytosis. Expert. Opin. Pharmacother., 6(14): 2435–2441.
21. Montella L., Insabato L., Palmieri G. (2004) Imatinib mesylate for cerebral Langerhans’-cell histiocytosis. N. Engl. J. Med., 351(10): 1034–1035.
22. Janku F., Amin H.M., Yang D. et al. (2010) Response of histiocytoses to imatinib mesylate: fire to ashes. J. Clin. Oncol., 28(31): e633–e636.
23. Ingram W., Desai S.R., Gibbs J.S., Mufti G. (2006) Reduced-intensity conditioned allogeneic haematopoietic transplantation in an adult with Langerhans’ cell histiocytosis and thrombocytopenia with absent radii. Bone Marrow Transplant., 37(7): 713–715.
24. Xicoy B., Ribera J.M., Batlle M., Feliu E. (2006) Sustained remission in an adult patient with Langerhans cell histiocytosis following T-cell depleted allogenic cell transplantation. Med. Clin. (Barc), 127(18): 716 (in Spanish).
25. Cassady J.R. (1987) Current role of radiation therapy in the management of histiocytosis-X. Hematol. Oncol. Clin. North Am., 1(1): 123–129.
26. Heyd R., Strassmann G., Donnerstag F. et al. (2000) Radiotherapy in Langerhans-cell histiocytosis. 2 case reports and review of the literature. Rontgenpraxis, 53(2): 51–61 (in German).
27. Micke O., Seegenschmiedt M.H. (2002) Consensus guidelines for radiation therapy of benign diseases: a multicenter approach in Germany. Int. J. Radiat. Oncol. Biol. Phys., 52(2): 496–513.
Адрес:
Крячок Ирина Анатольевна
03022, Киев, ул. Ломоносова, 33/43
Национальный институт рака
Тел.: (044) 257-10-90





Leave a comment