




Вторинний системний AL-амілоїдоз, пов’язаний з множинною мієломою: клінічний випадок і огляд літератури
Рудюк Т.О.1, Новосад О.І.2, Шевчук Л.А.2, Кундіна В.В.3, Непомнящий В.М.4, Шмідт А.А.5, Крячок І.А.2
- 1Національний медичний університет імені Богомольця, Київ
- 2Національний інститут раку, Київ
- 3Національний університет охорони здоров’я України імені П.Л. Шупика, Київ
- 4Інститут нефрології Національної академії медичних наук України, Київ
- 5Інститут серця МОЗ України, Київ
Резюме. AL-амілоїдоз належить до системного типу захворювань і характеризується агрегацією неправильно згорнутого легкого ланцюга імуноглобуліну, переважно в серці та нирках, викликаючи органну недостатність. У цій статті описується клінічний досвід ведення пацієнта з амілоїдозом серця, асоційований з множинною мієломою. Діагноз амілоїдозу ґрунтується на високому індексі клінічної підозри. На жаль, на ранніх стадіях перебіг хвороби часто є безсимптомним до прогресування аж до пізньої стадії, і навіть тоді симптоми можуть бути дуже неспецифічними. Тривалий час до встановлення діагнозу, часто більше 1 року, є частим фактором незадовільних результатів лікування. Сповільнений перебіг захворювання у більшості випадків призводить до фатальних наслідків, оскільки первинно пацієнти не звертаються до профільних фахівців. Застійна серцева недостатність та серцева аритмія є частими причинами смерті від первинного системного амілоїдозу, причиною 48–65% смертей є серцево-судинні прояви. Більш точне розпізнавання AL-амілоїдозу кардіологами дозволяє призначити якомога раніше лікування і поліпшити його результати. Традиційне лікування пацієнтів з множинною мієломою та AL-амілоїдозом включає комбінацію дексаметазону з бортезомібом та циклофосфамідом. Трансплантація гемопоетичних стовбурових клітин після прийому високих доз мелфалану стала ще одним варіантом лікування, що сприяє ремісії. Представлено клінічний випадок своєчасної повної діагностики і лікування комбінації цих двох захворювань, у результаті чого у пацієнта досягнуто повної гематологічної та часткової органної відповіді за основним захворюванням.
DOI: 10.32471/clinicaloncology.2663-466X.41-1.28050
Одержано: 21.09.2021
Прийнято до друку: 27.09.2021
Клінічний випадок
З огляду на рідкісність патології, практичний інтерес представляє клінічний випадок пацієнта В., 1959 року народження, який вперше в травні 2020 р. звернувся до Національного інституту фтизіатрії і пульмонології імені Ф.Г. Яновського зі скаргами на часту задишку і раптову втрату свідомості, у результаті якої отримав сильний забій правої половини грудної клітки. На оглядовій рентгенограмі органів грудної клітки (ОГК) було виявлено збільшені ліві відділи серця і правобічний ексудативний плеврит.
Хворого було направлено на консультацію до Національного інституту серцево-судинної хірургії імені М.М. Амосова, де за результатами ехокардіографії (Ехо-КГ) підтвердилося збільшення і розширення лівих відділів серця, ущільнення стулок та інфільтрація міокарда. Однак скорочувальна здатність міокарда лівого шлуночка (ЛШ) при цьому зберігалася на рівні 55%. Кардіолог вперше запідозрив діагноз: гіпертрофічна кардіоміопатія, необструктивна форма (?), амілоїдоз серця(?). А також у пацієнта зареєстрували персистуючу форму фібриляції передсердь і полісерозит. Хворий отримав симптоматичну терапію, і його направили на дообстеження.
Через 2 тиж пацієнта у важкому стані госпіталізовано в міську клінічну лікарню. Приводом для цього стала раптова втрата свідомості. З підозрою на амілоїдоз серця хворого направили на магнітно-резонансну томографію (МРТ) серця з в/в контрастуванням препаратом гадобутролу.
МРТ-ознаки кардіального амілоїдозу підтвердилися за ознаками наявності концентричної бівентрикулярної гіпертрофії без дилатації, гіпертрофії міжпередсердної перегородки, тенденції до дилатації передсердь, потовщення стулок клапанів і стінок передсердь.
До моменту надходження в Національний інститут раку (НІР) для лікування основного захворювання, а саме амілоїдозу серця, асоційованого з множинною мієломою (ММ), минуло 2 міс. З анамнезу відомо, що найпершими передвісниками амілоїдозу з ураженням серця були відчуття перебоїв у роботі серця, задишка і випадки втрати свідомості з’явилися у пацієнта ще в квітні 2020 р. Діагноз амілоїдозу ґрунтується на високому індексі клінічної підозри. На жаль, перебіг хвороби часто є безсимптомним до розвитку пізньої стадії захворювання, і навіть тоді симптоми можуть бути дуже неспецифічними.
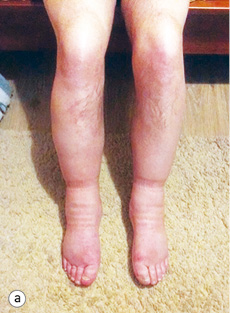
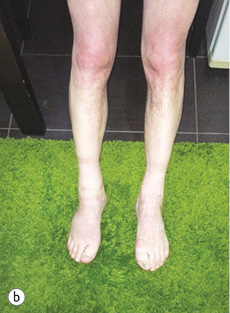
Уже під час звернення до НІР у пацієнта спостерігалася анасарка, набряки на ногах до гомілковоступневого суглоба (рис. 1 а, b).
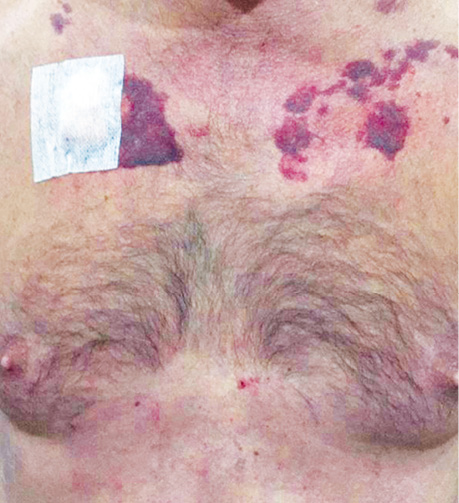
На момент госпіталізації була наявна периорбітальна пурпура, а також спостерігалися крововиливи на передній верхній третині грудної клітки (pис. 2).
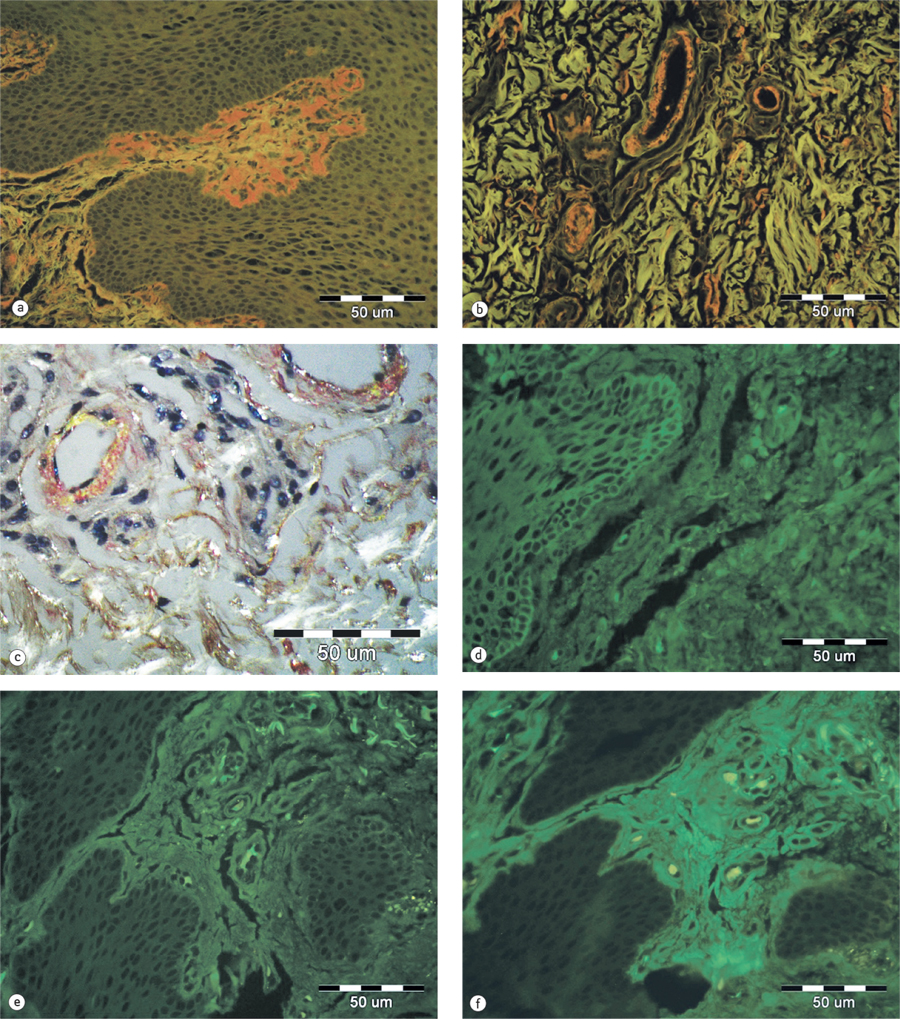
Під час додаткового обстеження, згідно з міжнародними критеріями діагностики AL-амілоїдозу, виявлено збільшену кількість плазматичних клітин у кістковому мозку, високий рівень легких ланцюгів імуноглобулінів у сироватці крові та сечі, а також специфічні маркери амілоїдозу, однак важких ланцюгів в сироватці крові і сечі не виявлено (табл. 1) [1–2].
Таблиця 1. Лабораторні показники пацієнта В. на всіх етапах лікування
| Показники | До лікування | Після 3 курсів специфічного лікування | Після 6 курсів специфічного лікування |
| NT-proBNT | 4200 пг/мл | 2293 пг/мл | 2911 пг/мл |
| Тропонін I | 0,17 нг/мл | <0,1 нг/мл | <0,1 нг/мл |
| Аналіз на легкі ланцюги в сироватці крові | Каппа — 15,8, лямбда — 1050, співвідношення: 0,02 | Каппа — 9, лямбда — 24, співвідношення: 0,38 | Каппа — 25,3, лямбда — 15,6, співвідношення: 1,62 |
| Аналіз на легкі ланцюги в сечі | Не виявлено | Не виявлено | Не виявлено |
| β2-мікроглобулін | 12 мг/л | 4,51 мг/л | 2,62 мг/л |
| Плазматичні клітини | 47% | 2% | 0,4% |
Діагностика системного амілоїдозу зазвичай вимагає гістологічного підтвердження, коли під час фарбування конго червоним виявляють відкладення амілоїдозу, яке візуалізується у вигляді зеленого подвійного променезаломлення при перегляді в крос-поляризованому світлі.
Пацієнту В. було проведено біопсію слизової оболонки щоки, за результатами якої і було встановлено діагноз AL-амілоїдозу (рис. 3).
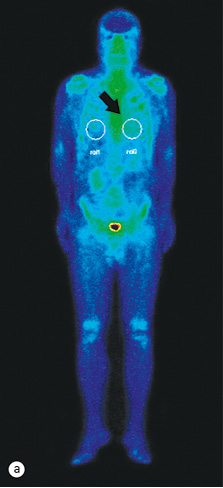
Для визначення обсягу ураження серцевого м’яза амілоїдними структурами і оцінки ефективності проведеного лікування цьому хворому з ознакою рестриктивної кардіоміопатії було проведено радіонуклідне дослідження — планарну сцинтиграфію тіла (Infinia Hawkeye, 2,0 мЗв) та однофотонну емісійну комп’ютерну томографію грудної клітки (рис. 4 а, b).
Для виявлення патологічної фіксації в ділянці серця використовують радіофармпрепарат і на підставі вимірювання показника R (співвідношення ROI 0/1) встановлюють інтенсивність ураження досліджуваної ділянки.
До лікування коефіцієнт накопичення R=ROI 0/ROI 1 у пацієнта становив 1,25, що відповідало 1-му ступеню ураження (рис. 4 а) і характерно для амілоїдних структур — легких ланцюгів. Показник ROI 0 — кількість гамма-Каунті, отриманої від ділянки серця (вказано стрілкою), показник ROI 1 — кількість гамма-Каунті, отриманої від контралатеральної ділянки серця (рис. 4b).
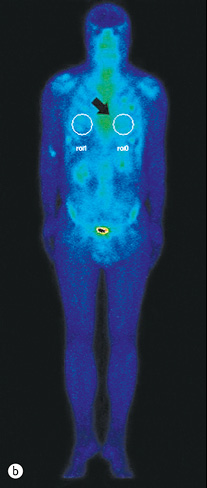
Після лікування виявлено зниження коефіцієнта накопичення радіофармпрепарату в ділянці серця до 1,13, що відповідає значному поліпшенню скорочувальної функції серця.
На підставі перерахованих вище результатів дослідження пацієнту встановили діагноз: ММ, типу IgG/lambda, II стадія за ISS. Вторинний системний AL-амілоїдоз з ураженням серця, III стадія.
Супутні захворювання: персистуюча форма фібриляції передсердь, тахісистолічний варіант, СНА2DS2-VASc — 2Б. HAS BLED — 3Б. EHRA III ст. Гіпертонічна хвороба II ступеня, 3 ст., ризик 4 (дуже високий). Серцева недостатність (СН) ІІА ст. Гостре пошкодження нирок, стадія ІІ AKIN.
За рішенням консиліуму план лікування складався з проведення 6 курсів ПХТ за схемою VCd (бортезоміб + циклофосфамід + дексаметазон) з подальшою аутологічною трансплантацією гемопоетичних стовбурових клітин (аутоСКПК) при досягненні позитивного результату лікування. Після 6-го курсу у хворого досягнуто повну гематологічну відповідь і часткову органну відповідь згідно з критеріями амілоїдозу.
У січні 2021 р. проведено високодозову хіміотерапію з подальшою аутоСКПК. Також пацієнт з моменту початку курсу специфічного лікування і до сьогодні знаходиться під наглядом кардіолога і отримує профілактичну антидіуретичну і кардіологічну терапію.
На даний момент хворий знаходиться в ремісії і наступний візит для оцінки ефективності лікування заплановано на + 100-й день.
Обговорення
Амілоїдоз належить до групи рідкісних захворювань, що характеризується позаклітинним відкладенням неправильно згорнутих білкових фібрил.
Захворюваність на AL-амілоїдоз в західних країнах становить 1 випадок на 100 000 населення. У Сполучених Штатах реєструється 1275–3200 нових випадків на рік. Щорічна частка нових випадків AL-амілоїдозу становить 78%. Згідно з даними Національної організації з рідкісних захворювань (National Organization for Rare Disorders — NORD), щорічно в США реєструються близько 4000 нових випадків захворювання на амілоїдоз легких ланцюгів (AL), що становить приблизно 8–12 осіб на 1 млн [3–5]. Незважаючи на значну смертність від амілоїдозу, публікацій стосовно даного захворювання досить мало. Наприклад, фінські колеги повідомляють, що смертність від АА-амілоїдозу, що ускладнює ревматоїдний артрит, становить 9–24% [6]. В іспанському 9-річному дослідженні у пацієнтів з ревматоїдним артритом зареєстровано 17% смертей, які пов’язані з амілоїдозом [7]. Навпаки, в інших дослідженнях смертність від вторинного амілоїдозу була набагато нижчою 2–7% [8].
Частота захворюваності на амілоїдоз у Європі значно нижча. Наприклад, в Англії на 1 млн осіб відмічаються до 3 випадків AL-амілоїдоз і 1 випадок АА-типу. Статистичні дані Швеції можна порівняти з даними Англії (3:2) [9].
На жаль, в Україні статистика щодо захворюваності та поширеності AL-амілоїдозу відсутня.
У зв’язку з поліпшенням матеріально-технічної бази лікарень і підвищенням кваліфікації лікарів на сьогодні в Україні почали проводити своєчасне дообстеження пацієнтів та їх лікування. Однак не проводилися ретро- або проспективні дослідження щодо цього захворювання і, відповідно, опубліковані дані малочисельні, враховуючи також те, що хвороба має уповільнений характер.
Виділяють амілоїдоз легких ланцюгів імуноглобуліну (AL), амілоїдоз білка амілоїду A (AA), спадковий транстиретиновий (Hereditary ATTR amyloidosis) і амілоїдоз дикого типу (Wild-type ATTR amyloidosis) (табл. 2). Ці типи амілоїдозу виникають у результаті впливу генетичних факторів, запальних захворювань і віку відповідно [10]. Локалізований AL-амілоїдоз відмічають рідко, він становить приблизно 7–8% випадків амілоїдозу [11].
Таблиця 2. Характеристики різних типів амілоїдозу
| Тип | Білок-попередник | Ураження основних органів | Діагностика | Лікування | Специфічні характеристики |
| Амілоїдоз легких ланцюгів, первинний | Амілоїд легкого ланцюга | Нирки
Серце Центральна нервова система Гастроінтестинальна пухлина (шлунково-кишковий тракт) Печінка Шкіра |
Імунофіксація білка сироватки та сечі
Мієлограма (5–10% плазматичних клітин) Біопсія підшкірної жирової клітковини Біопсія кісткового мозку |
Хіміотерапія
Трансплантація стовбурових клітин |
Співвідношення чоловіки:жінки — 3:2
10–15% пов’язано з множинною мієломою |
| Амілоїдоз білка амілоїду А | Амілоїд А | Нирки | – | Лікування основного захворювання | – |
| Сеніальний системний амілоїдоз | Спадковий транстиретин, амілоїдоз дикого типу | Серце
Тунельний синдром зап’ястя |
99m сцинтиграфія Tc-DPD Генетичне дослідження транстиретину
Лінійні ендокардіальні відкладення |
– | Співвідношення чоловіки:жінки — 20:1 |
| Спадковий транстиретиновий амілоїдоз (аутосомно-домінантний) | Транстиретин, мутований | Серце
Печінка Центральна або периферична нервова система |
99m сцинтиграфія Tc-DPD Генетичне дослідження транстиретину | Трансплантація
печінки Тафамідіс |
– |
| Ізольований амілоїдоз передсердь | Натрійуретичний пептид передсердь | – | – | – | Зростання захворюваності з віком та у жінок |
30% пацієнтів з вперше встановленим діагнозом ММ мають приховані відкладення амілоїду в кістковому мозку без ураження внутрішніх органів [12]. Ці відкладення можуть не мати ніякого відношення до звичайного перебігу і симптоматики ММ, але симптоматичний амілоїдоз в кінцевому підсумку може розвинутись у 12–15% таких пацієнтів [13].
Приблизно у 8% пацієнтів з AL-амілоїдозом може бути наявна супутня ММ з ознаками активного захворювання згідно з CRAB-критеріями (гіперкальціємія, ниркова недостатність, анемія, ураження кісток) [14].
Первинний AL-амілоїдоз рідко трансформується в активну MM [15]. У пацієнти з моноклональною гаммапатією невизначеного генезу (MGUS) відмічають у вісім разів вищий відносний ризик розвитку AL-амілоїдозу [16].
При AL-амілоїдозі і ММ виявляють патологічно змінені плазматичні клітини із загальними генетичними абераціями і частково збігаються клінічні симптоми, все ж це може бути два окремих захворювання. До того ж AL-амілоїдоз має відмінний від мієломи профіль експресії генів і прогностичних факторів [17].
Патогенез. Критерії встановлення діагнозу
На сьогодні відомо більше 36 різних білків-попередників амілоїду, кожен з яких має тропізм до різних органів-мішеней [18].
Структура амілоїду утворюється при взаємодії з каркасними білками, такими як глікозаміноглікани, амілоїдний білок сироватки та аполіпопротеїни [19]. У кінцевому рахунку утворюється аморфне нерозчинне відкладення, стійке до руйнування.
Діагноз встановлюється на підставі імуногістохімічного дослідження біопсії ураженого органу/тканини. Амілоїдні відкладення зазвичай можуть бути ідентифіковані при біопсії уражених органів, неінвазивній «скринінговій» біопсії віддають альтернативу, включаючи аспірат абдомінального жиру, що може ідентифікувати відкладення амілоїду у 60–80% пацієнтів із системним AL-амілоїдозом, і рідше — з типом ATTR [20].
Біопсія серця є найбільш точним діагностичним доказом амілоїдної кардіоміопатії, і, якщо діагноз не підтверджується біопсією іншої тканини, проводять ендоміокардіальну біопсію, яка є безпечною і відносно простою процедурою для кваліфікованого спеціаліста.
При системному амілоїдозі попередники легких ланцюгів імуноглобуліну (найчастіше лямбда-ізотип) продукуються в кістковому мозку. Далі циркулюючі легкі ланцюги відкладаються практично в будь-якій тканині, що призводить до дисфункції органу і в подальшому — систем органів.
Чи не найважливішим для підтвердження діагнозу, типу амілоїдозу та стадіювання захворювання є визначення біомаркерів серцевого і ренального ураження в крові (N-кінцевий пронатрійуретичний пептид тип-B (NT-proBNP), тропонін Т і/або I) і в сечі (добова протеїнурія), а також проведення інструментальних методів дослідження (ехокардіографії, МРТ з контрастом гадобутролом) і генетичних тестів.
Амілоїдоз серця
Ранній кардіальний амілоїдоз — серйозна діагностична проблема. Класичні ознаки «правобічної» СН можуть не проявлятись доти, поки серцеве захворювання не буде декомпенсованим. Ураження серця є основною причиною смерті при системному амілоїдозі, незалежно від патогенезу, що лежить в основі продукції амілоїду [21].
Інфільтративний процес призводить до потовщення стінки двох шлуночків з концентричним ремоделюванням їх та низьким серцевим викидом. Подальше підвищення тиску в передсердях пов’язане з дилатацією передсердь. Інтраміокардіальні судини часто інфільтруються амілоїдом, що викликає зниження перфузії міокарда [22].
Часто порушується провідна система серця, що призводить до розвитку передсердної аритмії (фібриляція, тріпотіння або передсердна тахікардія) і затримки атріовентрикулярної провідності.
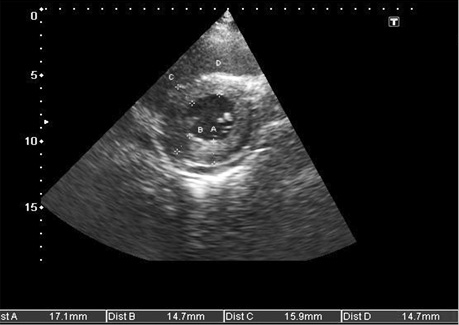
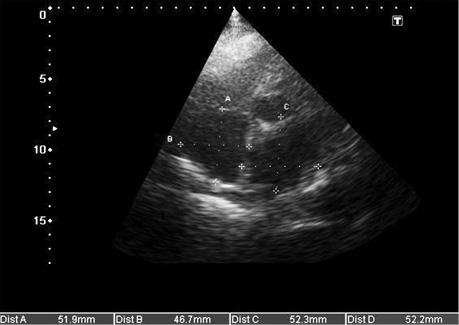
З урахуванням характеру захворювання, відсутності специфічних патогномонічних симптомів хвороби, провідне місце в діагностиці амілоїдозу серця займає ехо-КГ і допплерехокардіографія (допплер-ехо-КГ) [23].
Згідно з даними C. Rapezzi і співавторів, оцінка результатів ехо-КГ залежить від типу амілоїдозу і, отже, ступеня ураження шлуночків серця [24].
Відомо, що ехографічна оцінка є схожою між типами AL і ATTR-амілоїдозом, хоч їм і притаманні деякі відмінності. Наприклад, гіпертрофія лівого шлуночка, як правило, є симетричною при AL-амілоїдозі, а при ATTR спостерігається асиметрична сигмовидна гіпертрофія перегородки [25].
ATTR-тип характеризується значним збільшенням маси лівого шлуночка в порівнянні з такою правого [26].
Крім того, у пацієнтів з wtATT-типом відмічають більш виражене збільшення маси лівого шлуночка і більше зниження фракції викиду лівого шлуночка.
На жаль, на початкових етапах захворювання, при проведенні ехо-КГ, досить часто роблять помилковий висновок про наявність у пацієнта гіпертензивного серця або проявів гіпертрофічної ідіопатичної кардіоміопатії. Саме на етапі клінічної маніфестації амілоїдозу, розвитку проявів рефрактерної СН потрібно замислюватися про невідповідність клінічних проявів при гіпертензивному серці, гіпертрофічній ідіопатичній кардіоміопатії і сонографічній картині амілоїдного ураження серця [34].
З огляду на той факт, що амілоїдоз серця є проявом рестриктивної кардіоміопатії (РКМП), він сонографічно матиме сукупні ознаки: ехо-симптоми рестриктивної гемодинаміки серця і міокардіальної дисфункції з власне ехо-особливостями амілоїдозу [35].
Сонографічні характеристики амілоїдозу серця включають:
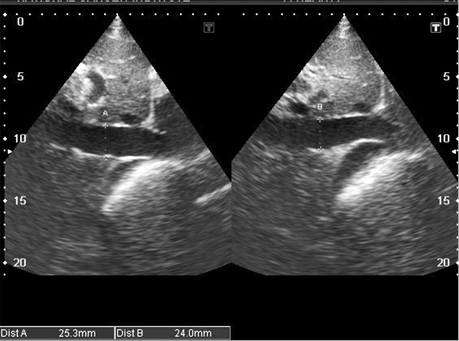
- збільшення кінцевого діастолічного потовщення міжшлуночкової перегородки більше 12 мм за відсутності інших причин гіпертрофії міокарда (артеріальна гіпертензія, аортальний стеноз) (рис. 5, 7, 8, 9);
- інфільтративне гомогенне потовщення, інфільтрація атріовентрикулярних клапанів, що супроводжується розвитком регургітації;
- дифузне інфільтративне потовщення інших стінок камер серця, включаючи передню стінку правого шлуночка, міжпередсердної перегородки (рис. 7, 8, 9);
- характерний блискучий «сальний», за типом «матового скла» ехо-сигнал за рахунок амілоїдної інфільтрації міокарда (рис. 9);
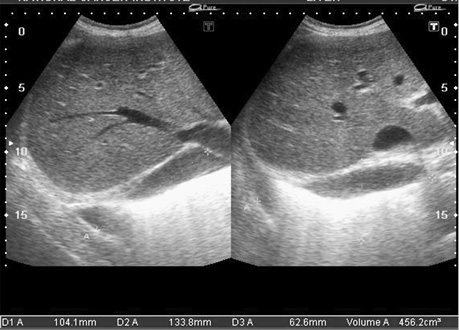
- дилатація передсердь, невідповідність їх порожнинам шлуночків (маленькі порожнини шлуночків за великих розмірів передсердь) (рис. 10);
- дилатація порожнистих, печінкових і легеневих вен, наявність вільної рідини в перикарді, плевральних порожнинах; (рис. 7, 11, 12);
- зниження швидкості кровотоку в печінкових венах на вдиху (рис. 11) і реверсія діастолічного потоку на видиху, властиві РКМП;
- у багатьох випадках зниження глобальної функції міокарда (фракція викиду ЛШ), порушення діастолічної функції (у тяжких випадках — рестриктивний тип діастолічної кривої) (рис. 9, 10).
Таким чином, основними сонографічними аспектами при амілоїдозі серця під час проведення ехо-КГ з допплер-ехо-КГ є визначення діастолічної дисфункції (порушення релаксації, псевдонормалізація, рестриктивний тип кривої) з перерахованими вище морфологічними порушеннями і ознаками інфільтративної РКМП, властивими їй порушеннями центральної гемодинаміки, порушенням кровотоку в легеневих і печінкових венах.
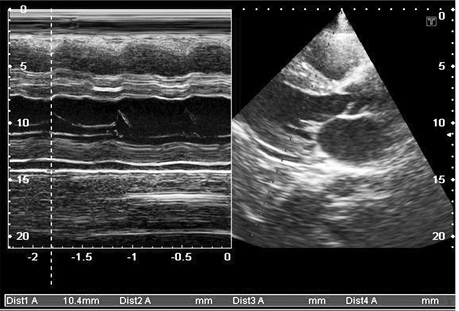
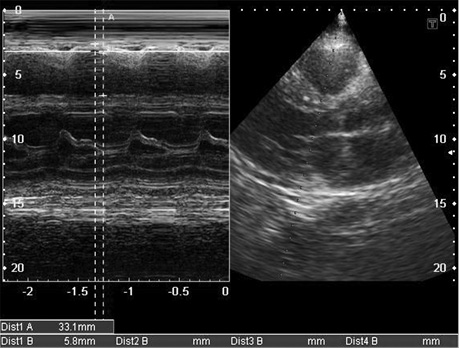
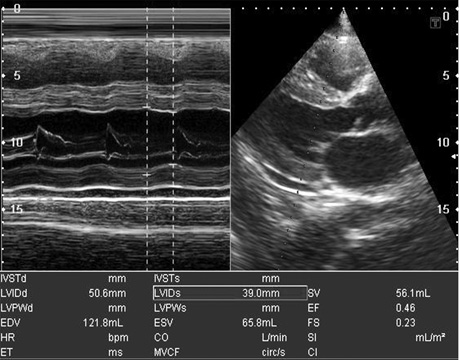
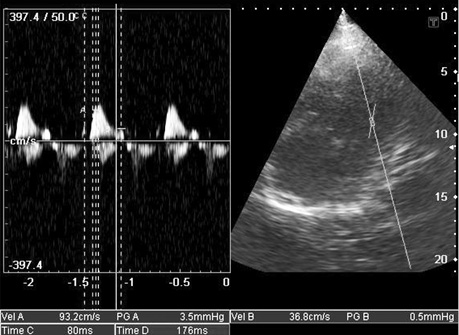
Крім ехокардіографії, існує ще один метод візуалізації — картування магнітного резонансу. Даний метод дозволяє детально досліджувати інтерстицій міокарда та оцінити характерні зміни з пізнім підвищенням рівня гадолінію (ПРГ) [27].
За допомогою ПРГ не можна розрізнити типи амілоїдозу AL і ATTR, але воно може допомогти в ранньому виявленні накопичення амілоїдозу в серцевому м’язі до появи явної гіпертрофії лівого шлуночка.
Повідомлялося про декілька різних моделей посилення при візуалізації ПРГ, проте найбільш поширені розподілення включають глобальні субендокардіальні і трансмуральні зміни [28].
Було виявлено також, що субендокардіальний ПРГ є переважаючим компонентом при AL-амілоїдозі, тоді як трансмуральний частіше відмічають при ATTR-типі [29].
Типовий компонент ПРГ має чутливість 85–90% для діагностики серцевого амілоїдозу. Візуалізація ПРГ також може використовуватися для моніторингу прогресування захворювання у міру прогресування амілоїдної інфільтрації, що дозволяє ПРГ служити незалежним предиктором прогнозу [30].
Ендоміокардіальна біопсія була золотим стандартом для підтвердження серцевого амілоїдозу, однак радіонуклідна сцинтиграфія з використанням міченого 99m-технецію стала високочутливим і дуже специфічним методом діагностики серцевого амілоїдозу ATTR-типу.
A. Castano, M. Haq та співавтори довели, що ступінь поглинання міокарда корелює із загальною смертністю [31].
Існують три основні часто використовувані індикатори, такі як: 99mTc-мічений пірофосфат (PYP), 3,3-дифосфоно-1,2-пропандикарбонова кислота (DPD) і гідроксиметилендифосфонат (HMDP), причому 99mTc-PYP є найбільш часто використовуваним індикатором у США.
На даний момент невідомо, чому в серці, ураженому ATTR, спостерігається більше поглинання 99mTc-PYP.
O.B. Suhr та співавтори припустили, що цей факт, можливо, пов’язаний з більш високим вмістом кальцію або складом амілоїдних фібрил при ATTR у порівнянні з іншими типами амілоїдозу [32].
Пацієнтам з позитивним результатом сканування 99mTc-PYP без моноклональних білків у крові і сечі може бути встановлено діагноз ATTR без тканинного підтвердження, враховуючи це, специфічність і прогностична цінність цього методу становить >98% [33].
Цінність цього дослідження полягає в можливості діагностувати амілоїдоз без тканинного підтвердження в окремих випадках, коли у пацієнтів існує СН, ехо-КГ або картування магнітного резонансу, які вказують на амілоїдоз, а також серцеве поглинання 2-го або 3-го ступеня при радіонуклідному скануванні та відсутність моноклонального білка.
Лікування
Ефективність лікування залежить від своєчасної діагностики. За останні три десятиліття результати лікування ММ значно покращилися і пов’язано це в першу чергу із застосуванням таргетних препаратів у комбінації з хіміопрепаратами або без, з подальшою консолідуючою терапією і аутоСКПК. Лікування пацієнтів з AL-амілоїдозом включає курси терапії, аналогічні тим, які застосовуються для пацієнтів з ММ.
До сьогодні оптимальною комбінацією терапії першої лінії для пацієнтів з вперше встановленим діагнозом AL-амілоїдозу був режим VCd (бортезоміб-циклофосфамід-дексаметазон).
Схема терапії даратумумаб-бортезоміб-циклофосфамід-дексаметазон (Dara-VCd) вперше зареєстрована Управлінням з контролю за харчовими продуктами і лікарськими засобами США (Food and Drug Administration — FDA) на початку 2021 р. за результатами дослідження ANDROMEDA і рекомендована для терапії першої лінії для лікування пацієнтів з первинним AL-амілоїдозом.
Група дослідників на чолі з Giovanni Palladini, продемонстрували не тільки стійку гематологічну і органну відповіді, але й задовільну переносимість режиму Dara-VCd. Результати лікування невеликої когорти пацієнтів (28 осіб) показали високий рівень загального гематологічного та дуже хорошу часткову відповіді (96 і 82% відповідно). Стійка органна відповідь спостерігалася майже у 50% пацієнтів [36].
Дискутабельним залишається питання тактики терапії в разі важкого ураження органів, оскільки проведення агресивної терапії стає неможливим.
Висновки
Представлений клінічний випадок показує, як важливо вчасно провести обстеження пацієнта і правильно скоригувати лікувальну тактику, незважаючи на коморбідність хворого та рідкісність поєднаної патології. Терапія пацієнтів з AL-амілоїдозом спрямована на лікування як основного захворювання, так і на зниження інтенсивності супутніх симптомів.
Конфлікт інтересів
Автори заявляють про відсутність конфлікту інтересів.
Дотримання прав пацієнтів і правил біоетики
Пацієнт підписав інформовану згоду на публікацію своїх даних.
Список використаної літератури
1. Gertz, M. A. (2018) Immunoglobulin light chain amyloidosis diagnosis and treatment algorithm. Blood Cancer Journal — Nature Publishing Group UK, 8: 44. doi: 10.1038/s41408-018-0080-9.
2. Gertz, M. A, & Kyle, R. A. (1989). Primary systemic amyloidosis a diagnostic primer. Mayo Clin Proc England. 64: 1505–19. doi: 10.1016/s0025-6196(12)65706-1.
3. Comenzo, R., Reece, D., Palladini, G., & Merlini, G. (2012). Consensus guidelines for the conduct and reporting of clinical trials in systemic light-chain amyloidosis. Leukemia, 26(11): 2317–2325. doi:10.1038/leu.2012.100.
4. Pinney, J., Smith, C., Taube, J., & Gillmore, J. (2013). Systemic Amyloidosis in England: an epidemiological study. British Journal of Haematology, 161(4): 525–532. doi:10.111/bjh.12286.
5. Laakso, M., Mutru, O., & Isomaki, H. (1986). Mortality from amyloidosis and renal diseases in patients with rheumatoid arthritis. Annals of the Rheumatic Diseases. 45: 663–667. doi: 10.1136/ard.45.8.663.
6. Martinez, M. S., Garcia-Monforte, A., & Rivera, J. (2001). Survival study of rheumatoid arthritis patients in Madrid (Spain). A 9-year pro-spective follow-up. Scandi Journal of Rheumatology. 30: 195–198. doi:10.1080/030097401316909512.
7. Kvalvik, A. G., Jones, M. A., & Symmons, D. P. (2000). Mortality in a cohort of Norwegian patients with rheumatoid arthritis followed from 1977 to 1992. Scandi Journal of Rheumatology. 29: 29–37. doi:10.1080/030097400750001770.
8. Pinney, J. H., Smith, C. J., Taube, J. B., & Gillmore Julian D. (2013, May). Systemic amyloidosis in England: an epidemiological study. British Journal of Haematology. 161(4): 525–32. doi:10.1111/bjh.12286.
9. Fernandes A., Caetano F., Almeida I., & Mota P. (2016). Amiloidose cardíaca — abordagem diagnóstica, a propósito de um caso clínico. Revista Portuguesa de Cardiologia. 35: 305.e1–305.e7. doi: 10.1016/j.repc.2016.01.004.
10. Sipe, J. D., Benson, M. D., Buxbaum J. N. & Per Westermark. (2010). Amyloid fibril protein nomenclature: recommendations from the nomenclature committee of the International Society of Amyloidosis Amyloid. The Journal of Protein Folding Disorders, 17: 3–4. doi: 10.3109/13506129.2010.526812.
11. Bahlis N. J., & Lazarus H. M. (2006). Multiple myeloma-associated AL amyloidosis: is a distinctive therapeutic approach warranted? Bone Marrow Transplant, 38: 7–15. doi:10.1038/sj.bmt.1705395.
12. Banypersad, S. M., Moon, J. C., Whelan, C. & Ashutosh D. Wechalekar. (23 Apr 2012). Updates in cardiac amyloidoses: A Review. Journal of the American Heart Association,1: 2. doi: 10.1161/JAHA.111.000364.
13. Kourelis T. V., Kumar S. K., Gertz M. A., & Hayman S. R. (2013). Coexistent multiple myeloma or increased bone marrow plasma cells define equally high-risk populations in patients with immunoglobulin light chain amyloidosis. Journal of Clinical Oncology (United State), 31: 4319–24. doi: 10.1200/JCO.2013.50.8499.
14. Rajkumar S. V, Gertz M. A, & Kyle R. A. (1998). Primary systemic amyloidosis with delayed progression to multiple myeloma. Cancer, 82(8): 1501–5. doi: 10.1002/(sici)1097-0142(19980415)82:8<1501::aid-cncr11>3.0.co;2-8.
15. Kyle R. A., Larson D. R., Therneau T. M., & Cerhan J. R. (January 18, 2018). Long-term follow-up of monoclonal gammopathy of undetermined significance. The New England Journal of Medicine, 378: 241–9. doi: 10.1056/NEJMoa179974.
16. Falk R. H. (2014). AL amyloidosis or multiple myeloma? An important distinction. British Journal of Haematology. John Wiley & Sons, ltd (10.1111).164: 748–9. doi: 10.1111/bjn.12677.
17. Desport E., Bridoux F., Sirac C., & Jaccard A. (2012). AL Amyloidosis. Orphanet Journal of Rare Diseases, 7(1): 54. doi: 10.1186/1750-1172-7-54.
18. Sanchorawala V. (2006). Light-Chain (AL) Amyloidosis: Diagnosis and Treatment. Clinical Journal of the American Society of Nephrology, 1(6): 1331–1341. doi:10.2215/CJN.02740806.
19. Guy C. D, Jones C. K. (2001). Abdominal fat pad aspiration biopsy for tissue confirmation of systemic amyloidosis: specificity, positive predictive value, and diagnostic pitfalls. Diagn Cytopathol. 24: 181–5. doi: 10.1002/1097-0339(200103)24:3<181::aid-dc1037>3.0.co;2-d.
20. Fontana M. Banypersad S. M. Treibel T. A. & Moon C. James. (2015). Differential myocyte responses in patients with cardiac transthyretin amyloidosis and light-chain amyloidosis: a cardiac MR imaging study. Radiology, 277:388–97. doi: 10.1148/radiol.2015141744.
21. Falk R. H. (2005). Diagnosis and management of the cardiac amyloidoses. Circulation, 112: 2047–60. doi: 10.1161/CIRCULATIONAHA.104.489187.
22. Koyama J., Ray-Sequin P. A. & Falk H.Rodney. (2002). Usefulness of pulsed tissue Doppler imaging for evaluating systolic and diastolic left ventricular function in patients with AL (primary) amyloidosis. The American Journal of Cardiology, 89: 1067–71. doi: 10.1016/s0002-9149(02)02277-4.
23. Claudio Rapezzi, Giampaolo Merlini & Perlini Stefano. (2009). Systemic cardiac amyloidoses: disease profiles and clinical courses of the 3 main types. Circulation. Nuances in the various subtypes of cardiac amyloidosis are highlighted, 120(13): 1203–12. doi: 10.1161/CIRCULATIOHANA.108.84334.
24. Martinez-Naharro A., Treibel T. A., & Knight D. S. (2017). Magnetic resonance in transthyretin cardiac amyloidosis. J Am Coll Cardiol, 70(4): 466–77. doi: 10.1016/j.jacc.2017.05.053.
25. Maurer M. S., Elliott P., Comenzo R., & Rapezzi C. (2017). Addressing common questions encountered in the diagnosis and management of cardiac amyloidosis. Circulation, 135(14): 1357–77. doi: 10.1161/CIRCULATIONAHA.116.024438.
26. Maceira A. M., Joshi J., Prasad S. K., & Harding I. (2005). Cardiovascular magnetic resonance in cardiac amyloidosis. Circulation, 111(2):186–93. doi: 10.1161/01.CIR.0000152819.97857.9D.
27. Dorbala Sharmila, Yukio Ando & Bourque M. Jamieson. (2019). ASNC/AHA/ASE/EANM/HFSA/ISA/SCMR/SNMMI expert consensus recommendations for multimodality imaging in cardiac amyloidosis: part 1 of 2-evidence base and standardized methods of imaging. Journal of Nuclear Cardiology, 26(6): 2065–123. doi: 10.1161/HCI.0000000000000029.
28. Fontana M., Pica S., Reant P., & Banypersad S. M. (2015). Prognostic value of late gadolinium enhancement cardiovascular magnetic resonance in cardiac amyloidosis. Circulation, 132(16): 1570–9. doi: 10.1161/CIRCULATIONAHA.115.016567.
29. Raina S., Lensing S. Y., Nairooz R. S., & Bhatti S. (2016). Prognostic value of late gadolinium enhancement CMR in systemic amyloidosis. JACC Cardiovasc Imaging, 9(11): 1267–77. doi: 10.1016/j.jcmg.2016.01.036.
30. Castano A., Haq M., Narotsky D. L., & Morgenstern R. (2016). Multicenter study of planar technetium 99m pyrophosphate cardiac imaging: predicting survival for patients with ATTR cardiac amyloidosis. JAMA Cardiology, 1(8): 880–9. doi: 10.1001/jamacardio.2016.2839.
31. Suhr O. B., Lundgren E., Westermark P.. (2017). One mutation, two distinct disease variants: unravelling the impact of transthyretin amyloid fibril composition. Journal of Internal Medicine, 281(4): 337–47. doi: 10.1111/joim.12585.
32. Gillmore J. D., Mauer S. Mathew & Hawkins N. Philip. (2016). Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation, 133(24): 2404–12. doi: 10.1161/CIRCULATIONAHA.116.021612.
33. Koyama J., Ray-Sequin P. A., Falk R. H. (2003). Longitudinal Myocardial Function Assessed by Tissue Velocity, Strain, and Strain Rate Tissue Doppler Echocardiography in Patients With AL (Primary) Cardiac Amyloidosis. Circulation, 107: 2446–2452. doi: 10.1161/01.CIR.0000068313.67758.4F.
34. М. Н. Долженко, С. В. Поташев, О. А. Шараевский. (2010). Констриктивный перикардит: клинический случай. Газета «Новости медицины и фармации». Кардиология, 327: 23–29.
35. Palladini Giovanni, Kastritis Efstathios & Comenzo L. Raymond. (2020). Daratumumab plus CyBorD for patients with newly diagnosed AL amyloidosis: safety run-in results of ANDROMEDA. Blood, 136(1): 71–80. doi: 10.1182/blood.2019004460.
Вторичный системный AL-амилоидоз, связанный с множественной миеломой: клинический случай и обзор литературы
1Национальный медицинский университет имени А.А. Богомольца, Киев
2Национальний институт рака, Киев
3Национальний университет здравоохранения Украины имени П. Л. Шупика, Киев
4Институт нефрологии АМН Украины, Киев
5Институт сердца МЗ Украины, Киев
Резюме. AL-амилоидоз относится к системному типу заболеваний и характеризуется агрегацией неправильно свернутой легкой цепи иммуноглобулина, преимущественно в сердце и почках, вызывая органную недостаточность. В этой статье описывается клинический опыт ведения пациента с амилоидозом сердца, ассоциированным с множественной миеломой. Диагноз амилоидоза основывается на высоком индексе клинического подозрения. К сожалению, на ранних стадиях болезнь часто протекает бессимптомно до прогрессирования до поздней стадии, и даже симптомы могут быть очень неспецифическими. Продолжительное время для установления диагноза, часто более 1 года, является частым фактором неудовлетворительных результатов лечения. Замедленное течение заболевания в большинстве случаев приводит к фатальным последствиям, так как первично пациенты не обращаются к профильным специалистам. Застойная сердечная недостаточность и сердечная аритмия являются частыми причинами смерти от первичного системного амилоидоза, 48–65% смертей случаются вследствие сердечно-сосудистых проявлений. Более точное распознавание AL-амилоидоза кардиологами позволяет назначить лечение как можно раньше и улучшить его результаты. Традиционная терапия пациентов с множественной миеломой и AL-амилоидозом включает комбинацию дексаметазона с бортезомибом и циклофосфамидом. Трансплантация гемопоэтических стволовых клеток после приема высоких доз мелфалана стала еще одним вариантом лечения, которое приводит к ремиссии. Представлен клинический случай своевременной полной диагностики и лечения комбинации этих двух заболеваний, в результате чего у пациента достигнуты полный гематологический и частичный органный ответы по основному заболеванию.
Ключевые слова: амилоидоз сердца; легкие цепи; даратумумаб; множественная миелома; конго красный
Адреса:
Новосад Ольга Ігорівна, Рудюк Тимур Олегович
03022, Київ, вул. Ломоносова, 33/43
Національний інститут раку
E-mail: novosad.o.ua@gmail.com, tymur.rudiuk@gmail.com
Correspondence:
Novosad Olga, Rudiuk Tymur
33/43 Lomonosova Str., Kyiv
National Cancer Institute
E-mail: novosad.o.ua@gmail.com, tymur.rudiuk@gmail.com





Leave a comment